Gene regulation. DNA is merely the blueprint Shared spatially (among all tissues) and temporally But cells manage to differentiate
|
|
|
- Meryl Pitts
- 5 years ago
- Views:
Transcription
1 Gene regulation DNA is merely the blueprint Shared spatially (among all tissues) and temporally But cells manage to differentiate Especially but not only during developmental stage And cells respond to external conditions and/or messages from other cells Much of this dynamic response is attained through protein or gene regulation: how much and which variant of the gene is present

2 The central dogma
3 Mechanisms of gene regulation Pre-transcription: accessibility of the gene the chromatin structure which packs the DNA is dynamic Transcription: rate Post-transcription: mrna degradation rate Translation: rate Post-translation: Modifications Rate of degradation

4 Transcription factors Bind to specific DNA sites: Transcription Factor Binding Sites Typically downstream effect on mrna transcription rate
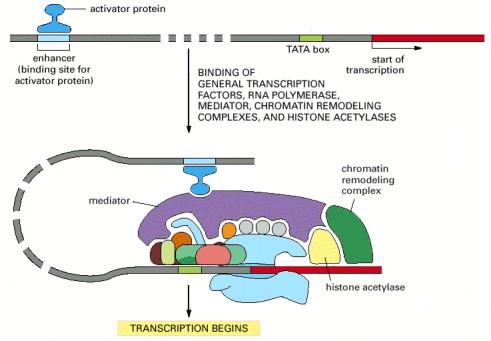

5 Transcription rate
6 Motif finding Motif finding is the computational problem of identifying TFBSs Implicit assumption: different TFBSs of the same TF should be similar to another Hence the name motif Two related tasks: Given a specific model of TF motif compiled from a known list of TFBSs find additional sites (scanning) Identify the unknown motif given only the DNA sequences
7 Modelling motifs Discovered sites: How do we model the motif? important for finding additional sites Consensus pattern: generalizes to regular expressions TACGAT TATAAT TATAAT GATACT TATGAT TATATT TATAAT Positional profile: A C 1 1 G 1 2 T
8 Generative models Consensus pattern: each instance is a randomly mutated version of the consensus substitution only: the same TF binds to the various sites, so indels are unlikely to occur as the DNA-TF contact region remains the same Profile: instances are drawn according to the probability implied by the positional profile assuming each position is drawn independently Pseudocounts are typically added to avoid excluding unseen letters
9 Counts to frequencies profile A C 1 1 G 1 2 T A C G T What is the pseudocount in this example?
10 The fitness of a TFBS 1 How well does a putative TFBS w fits the model? For a consensus model we typically use s C (w) = d H (C, w), the Hamming distance to the consensus pattern C. It is convenient to work with but more appropriate for uniform nucleotide sample For a profile parametrized by M = (f ik ) i=1:l,k=1:4, it is natural to use the likelihood score: s M (w) = P M (w) = l i=1 f iw i Better: use the LLR (loglikelihood ratio) score s M (w) = log P M(w) P B (w) = l i=1 log f iw i b wi, where B specifies an iid background model with nucleotide frequency (b k ) 4 1, typically taken from the organism or the scanned sample
11 Scanning for TFBS 2 Given a parametrized motif model and an associated fitness function looking for additional sites is algorithmically trivial However, setting a cutoff score typically requires carefully analyzing the FP rates These FP rates are set using a model of random sequences Markov chains shuffling using random chunks of DNA
12 Motif finding 3 Do these sequences share a common TFBS? tagcttcatcgttgacttctgcagaaagcaagctcctgagtagctggccaagcgagc tgcttgtgcccggctgcggcggttgtatcctgaatacgccatgcgccctgcagctgc tagaccctgcagccagctgcgcctgatgaaggcgcaacacgaaggaaagacgggacc agggcgacgtcctattaaaagataatcccccgaacttcatagtgtaatctgcagctg ctcccctacaggtgcaggcacttttcggatgctgcagcggccgtccggggtcagttg cagcagtgttacgcgaggttctgcagtgctggctagctcgacccggattttgacgga ctgcagccgattgatggaccattctattcgtgacacccgacgagaggcgtccccccg gcaccaggccgttcctgcaggggccaccctttgagttaggtgacatcattcctatgt acatgcctcaaagagatctagtctaaatactacctgcagaacttatggatctgaggg agaggggtactctgaaaagcgggaacctcgtgtttatctgcagtgtccaaatcctat
13 If only life could be that simple 4 The binding sites are almost never exactly the same A more likely sample is: tagcttcatcgttgacttttgaagaaagcaagctcctgagtagctggccaagcgagc tgcttgtgcccggctgcggcggttgtatcctgaatacgccatgcgccctggagctgc tagaccctgcagccagctgcgcctgatgaaggcgcaacacgaaggaaagacgggacc agggcgacgtcctattaaaagataatcccccgaacttcatagtgtaatctgcagctg ctcccctacaggtgcaggcacttttcggatgctgcttcggccgtccggggtcagttg cagcagtgttacgcgaggttctacagtgctggctagctcgacccggattttgacgga CTGCAGccgattgatggaccattctattcgtgacacccgacgagaggcgtccccccg gcaccaggccgttcctacaggggccaccctttgagttaggtgacatcattcctatgt acatgcctcaaagagatctagtctaaatactacctacagaacttatggatctgaggg agaggggtactctgaaaagcgggaacctcgtgtttatttgcattgtccaaatcctat
14 Searching for motifs 5 Simultaneously looking for a motif model and sites that will optimize a scoring function is significantly more difficult Assume for simplicity the OOPS model (One Occurrence Per Sequence model): w m S m for m = 1 : n A natural way to score a putative combination of a motif M and sites (w m ) n 1 is by summing the fitness scores of all sites: s(m; w 1,..., w n ) := n m=1 s M (w m ) Thus, our goal is to search the joint space of motifs, M (consensus or profile), and alignments, w m S m, so as to optimize this score Fortunately, for both models this can be done sequentially so we do not have to optimize simultaneously over the alignment and the motif
15 Optimizing the motif or the alignment 6 Once we choose the alignment, w m S m for m = 1 : n, the optimal motif for that alignment is trivial For the consensus model it is a consensus word as it clearly minimizes the total distance to the words in the alignment For the profile model we find with a little more effort that the best model is the one which coincides with how we define a profile: f ik = n ik n, where n ik is the number of occurrence of the letter k at position i. Conversely, if we know the model we can find the optimal sites for the putative motif by linearly scanning the sequences Often a motif finder will combine both the motif s and the alignment s optimizations and indeed they are in some sense equivalent
16 Heuristic vs. guaranteed optimizations 7 Assume for now l is known (we can enumerate over possible ls) and let N m be the length of S m By considering all, roughly, n m=1 N m gapless alignments made of w m S m we are guaranteed to find the optimal alignment under both possible motif models Unfortunately, this number is prohibitively expensive for all but a few cases
17 Finding an optimal pattern 8 Consistent with our previous discussion under the OOPS model the score of a consensus word C is often the total distance: T D(C) := n m=1 d H (C, S m ) = n m=1 min d H(C, w ) w S m Problem: find a word C that minimizes the total distance Naive solution: enumerate all 4 l possible consensus words Complexity: O(4 l D) While this approach is feasible for a larger set of parameters than the one available for alignment enumeration it is still often too expensive
18 Heuristic approaches: Sample Driven 9 Most of the 4 l patterns we explore in the exhaustive enumeration have little to do with our sample Sample driven approach: compute T D(w) only for words w in the sample Complexity: O(D 2 ) where D = n m=1 N m is the size of the sample Analysis: fast but can miss the optimal pattern if it is missing from the sample More sophisticated methods were developed based on the sample driven approach
19 CONSENSUS - greedy profile search (Hertz & Stormo 99) 10 Assume the OOPS model and that l is given There is a version that does not assume l is given (WCONSEN- SUS) CONSENSUS Follows a greedy strategy looking first for the best alignment of just two sites: For each i j, and w S i, w S j compute the information content of the alignment made of w and w : I = l i=1 4 k=1 n ik log n ik/2 b k Keep the top q 2 alignments (matrices)
20 It then greedily adds one word at a time from the sequences that are not already represented in the alignment 11 Let m := 3 denote the number of sequences in the current alignments While m < n for each of([ the top ]) saved q m 1 alignments A of m 1 rows A compute I for all words w which come from sequences w that are not already in A keep the best q m alignments and set m := m + 1
21 MEME (Bailey & Elkan 94) 12 MEME: Multiple EM for Motif Elicitation the multiple part is for dealing with multiple motifs probabilistic generative model, deterministic algorithm Recall that given the motif model we can linearly scan the sequences for instances Conversely, given the instances deducing the profile is trivial MEME alternates between the two tasks
22 MEME s outline 13 Starting from a heuristically chosen initial profile Sample driven: the profile is derived from the word in the sample that has a minimal total distance MEME iterates the following two steps until convergence score each word according to how well it fits the current profile update the profile by taking a weighted average of all the words The EM in MEME stands for Expectation Maximization (Dempster, Laird & Rubin 77) which MEME s two step procedure follows EM is guaranteed to monotonically converge to a local maximum (intelligent choice of a starting point is crucial)
23 Gibbs Sampler (Lawrence et al. 93) 14 Probabilistic framework, random algorithm Assumes the OOPS model for simplicity (many more variants) Suppose we selected putative instances w i S i, these define the profile or motif model as in MEME As in the EM context we compute the LR score of every word in the sample: L w = P M(w) P B (w) In EM we use a soft assignment of words to the list of selected sites (instances), alternatively we can use hard assignment: Iterate e.g., we can choose: w i = argmax w S i L w or, we can randomly choose a site with probability proportional to its LLR score (Gibbs Sampling)
24 Gibbs outline 15 Start with a random choice of w i S i While there has been an improvement in the total LLR score (over all sites) in the last L (the plateau period) iterations do For i = 1... n: remove word w i from the motif model M and randomly pick a new site from sequence S i with probability proportional to L w (an iteration is one such loop) Note that there is no convergence in the naive sense There is an alternative formulation in terms of a Gibbs Sampler: the goal is to randomly sample alignments from a distribution where each alignment s probability is roughly proportional to its LR score. The Gibbs Sampler defines an MCMC that converges to a stationary distribution with that property thus allowing us to sample from this distribution.
25 Is this significant? 16 Motif finders always find something A high scoring motif reported by CONSENSUS The alignment ccgataaggtaag TCGATAAGGaGAG TCGATAAaGTaAG TCGATAAGGTcAG TCGATAAGGTGAG The profile A C G T 4 5 4
26 Assessing the significance 17 How likely are we to see such alignments or better by chance? Clearly you need more information: What is the null model, or how are random (chance) sequences generated? typically iid (independent identically distributed or 0-th order Markov) What is the size of the search space: How many input sequences are there and how long are they? What was CONSENSUS instructed to look for? What is a better alignment, or how do we score a motif information content (Stormo 88): L i=1 A j=1 n ij log n ij/n b j
27 Quantifying the significance: the E-value / p-value 18 The E-value of an alignment with score s is: The expected number of random such alignments with score s... given the size of the search space An E-value of 0.01 is better than 100 It is computed by multiplying the size of the search space by the p-value of the alignment The p-value of an alignment with score s is: The probability that the score of a random alignment of the same width and depth is s
28 Phylogenetically aware finders 19 So far we looked at de novo motif finders: the only input is the set of presumably co-regulated sequences Binding sites are functional and functional elements tend to be more conserved Therefore we should look for conserved words in phylogenetically related species This can be done when we search for a known motif (MONKEY - Moses et al. 2004) Or, when looking for unknown motif (PhyloGibbs - Siddharthan et al. 2005) Other finders might add ChIP-chip data (MDscan - Liu et al. 2002)
Unsupervised Learning of Multiple Motifs in Biopolymers Using Expectation Maximization
 Machine Learning, 21, 51-80 (1995) 1995 Kluwer Academic Publishers, Boston. Manufactured in The Netherlands. Unsupervised Learning of Multiple Motifs in Biopolymers Using Expectation Maximization TIMOTHY
Machine Learning, 21, 51-80 (1995) 1995 Kluwer Academic Publishers, Boston. Manufactured in The Netherlands. Unsupervised Learning of Multiple Motifs in Biopolymers Using Expectation Maximization TIMOTHY
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Huber & Bulyk, BMC Bioinformatics MS ID , Additional Methods. Installation and Usage of MultiFinder, SequenceExtractor and BlockFilter
 Installation and Usage of MultiFinder, SequenceExtractor and BlockFilter I. Introduction: MultiFinder is a tool designed to combine the results of multiple motif finders and analyze the resulting motifs
Installation and Usage of MultiFinder, SequenceExtractor and BlockFilter I. Introduction: MultiFinder is a tool designed to combine the results of multiple motif finders and analyze the resulting motifs
Profiles and Multiple Alignments. COMP 571 Luay Nakhleh, Rice University
 Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
PROJECTION Algorithm for Motif Finding on GPUs
 PROJECTION Algorithm for Motif Finding on GPUs Jhoirene B. Clemente, Francis George C. Cabarle, and Henry N. Adorna Algorithms & Complexity Lab Department of Computer Science University of the Philippines
PROJECTION Algorithm for Motif Finding on GPUs Jhoirene B. Clemente, Francis George C. Cabarle, and Henry N. Adorna Algorithms & Complexity Lab Department of Computer Science University of the Philippines
Evolutionary tree reconstruction (Chapter 10)
") Evolutionary tree reconstruction (Chapter 10) Early Evolutionary Studies Anatomical features were the dominant criteria used to derive evolutionary relationships between species since Darwin till early
Evolutionary tree reconstruction (Chapter 10) Early Evolutionary Studies Anatomical features were the dominant criteria used to derive evolutionary relationships between species since Darwin till early
On the Optimality of the Neighbor Joining Algorithm
 On the Optimality of the Neighbor Joining Algorithm Ruriko Yoshida Dept. of Statistics University of Kentucky Joint work with K. Eickmeyer, P. Huggins, and L. Pachter www.ms.uky.edu/ ruriko Louisville
On the Optimality of the Neighbor Joining Algorithm Ruriko Yoshida Dept. of Statistics University of Kentucky Joint work with K. Eickmeyer, P. Huggins, and L. Pachter www.ms.uky.edu/ ruriko Louisville
Machine Learning. Computational biology: Sequence alignment and profile HMMs
 10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
Mixture Models and EM
 Mixture Models and EM Goal: Introduction to probabilistic mixture models and the expectationmaximization (EM) algorithm. Motivation: simultaneous fitting of multiple model instances unsupervised clustering
Mixture Models and EM Goal: Introduction to probabilistic mixture models and the expectationmaximization (EM) algorithm. Motivation: simultaneous fitting of multiple model instances unsupervised clustering
Chapter 2. Related Work
 Chapter 2 Related Work There are three areas of research highly related to our exploration in this dissertation, namely sequential pattern mining, multiple alignment, and approximate frequent pattern mining.
Chapter 2 Related Work There are three areas of research highly related to our exploration in this dissertation, namely sequential pattern mining, multiple alignment, and approximate frequent pattern mining.
Biology 644: Bioinformatics
 A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
Reddit Recommendation System Daniel Poon, Yu Wu, David (Qifan) Zhang CS229, Stanford University December 11 th, 2011
 Zhang CS229, Stanford University December 11 th, 2011") Reddit Recommendation System Daniel Poon, Yu Wu, David (Qifan) Zhang CS229, Stanford University December 11 th, 2011 1. Introduction Reddit is one of the most popular online social news websites with millions
Reddit Recommendation System Daniel Poon, Yu Wu, David (Qifan) Zhang CS229, Stanford University December 11 th, 2011 1. Introduction Reddit is one of the most popular online social news websites with millions
Software and Hardware Acceleration of the Genomic Motif Finding Tool PhyloNet
 Department of Computer Science & Engineering 2008-18 Software and Hardware Acceleration of the Genomic Motif Finding Tool PhyloNet Authors: Justin Brown Corresponding Author: jtb1@cec.wustl.edu Type of
Department of Computer Science & Engineering 2008-18 Software and Hardware Acceleration of the Genomic Motif Finding Tool PhyloNet Authors: Justin Brown Corresponding Author: jtb1@cec.wustl.edu Type of
Dynamic Programming Part I: Examples. Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, / 77
 Dynamic Programming -class4- July 25th, / 77") Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Gene expression & Clustering (Chapter 10)
") Gene expression & Clustering (Chapter 10) Determining gene function Sequence comparison tells us if a gene is similar to another gene, e.g., in a new species Dynamic programming Approximate pattern matching
Gene expression & Clustering (Chapter 10) Determining gene function Sequence comparison tells us if a gene is similar to another gene, e.g., in a new species Dynamic programming Approximate pattern matching
Finding Hidden Patterns in DNA. What makes searching for frequent subsequences hard? Allowing for errors? All the places they could be hiding?
 Finding Hidden Patterns in DNA What makes searching for frequent subsequences hard? Allowing for errors? All the places they could be hiding? 1 Initiating Transcription As a precursor to transcription
Finding Hidden Patterns in DNA What makes searching for frequent subsequences hard? Allowing for errors? All the places they could be hiding? 1 Initiating Transcription As a precursor to transcription
CLUSTERING. JELENA JOVANOVIĆ Web:
 CLUSTERING JELENA JOVANOVIĆ Email: jeljov@gmail.com Web: http://jelenajovanovic.net OUTLINE What is clustering? Application domains K-Means clustering Understanding it through an example The K-Means algorithm
CLUSTERING JELENA JOVANOVIĆ Email: jeljov@gmail.com Web: http://jelenajovanovic.net OUTLINE What is clustering? Application domains K-Means clustering Understanding it through an example The K-Means algorithm
On the Parameter Estimation of the Generalized Exponential Distribution Under Progressive Type-I Interval Censoring Scheme
 arxiv:1811.06857v1 [math.st] 16 Nov 2018 On the Parameter Estimation of the Generalized Exponential Distribution Under Progressive Type-I Interval Censoring Scheme Mahdi Teimouri Email: teimouri@aut.ac.ir
arxiv:1811.06857v1 [math.st] 16 Nov 2018 On the Parameter Estimation of the Generalized Exponential Distribution Under Progressive Type-I Interval Censoring Scheme Mahdi Teimouri Email: teimouri@aut.ac.ir
Clustering. CS294 Practical Machine Learning Junming Yin 10/09/06
 Clustering CS294 Practical Machine Learning Junming Yin 10/09/06 Outline Introduction Unsupervised learning What is clustering? Application Dissimilarity (similarity) of objects Clustering algorithm K-means,
Clustering CS294 Practical Machine Learning Junming Yin 10/09/06 Outline Introduction Unsupervised learning What is clustering? Application Dissimilarity (similarity) of objects Clustering algorithm K-means,
Gene Clustering & Classification
 BINF, Introduction to Computational Biology Gene Clustering & Classification Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Introduction to Gene Clustering
BINF, Introduction to Computational Biology Gene Clustering & Classification Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Introduction to Gene Clustering
I How does the formulation (5) serve the purpose of the composite parameterization
 serve the purpose of the composite parameterization") Supplemental Material to Identifying Alzheimer s Disease-Related Brain Regions from Multi-Modality Neuroimaging Data using Sparse Composite Linear Discrimination Analysis I How does the formulation (5)
Supplemental Material to Identifying Alzheimer s Disease-Related Brain Regions from Multi-Modality Neuroimaging Data using Sparse Composite Linear Discrimination Analysis I How does the formulation (5)
Framework for Design of Dynamic Programming Algorithms
 CSE 441T/541T Advanced Algorithms September 22, 2010 Framework for Design of Dynamic Programming Algorithms Dynamic programming algorithms for combinatorial optimization generalize the strategy we studied
CSE 441T/541T Advanced Algorithms September 22, 2010 Framework for Design of Dynamic Programming Algorithms Dynamic programming algorithms for combinatorial optimization generalize the strategy we studied
Sequence clustering. Introduction. Clustering basics. Hierarchical clustering
 Sequence clustering Introduction Data clustering is one of the key tools used in various incarnations of data-mining - trying to make sense of large datasets. It is, thus, natural to ask whether clustering
Sequence clustering Introduction Data clustering is one of the key tools used in various incarnations of data-mining - trying to make sense of large datasets. It is, thus, natural to ask whether clustering
Important Example: Gene Sequence Matching. Corrigiendum. Central Dogma of Modern Biology. Genetics. How Nucleotides code for Amino Acids
 Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Genome 559: Introduction to Statistical and Computational Genomics. Lecture15a Multiple Sequence Alignment Larry Ruzzo
 Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
3.4 Multiple sequence alignment
 3.4 Multiple sequence alignment Why produce a multiple sequence alignment? Using more than two sequences results in a more convincing alignment by revealing conserved regions in ALL of the sequences Aligned
3.4 Multiple sequence alignment Why produce a multiple sequence alignment? Using more than two sequences results in a more convincing alignment by revealing conserved regions in ALL of the sequences Aligned
Fitting a mixture model by expectation maximization to discover. Timothy L. Bailey and Charles Elkan y. and
 From: Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology August, 1994, pp. 28-36. AAAI Press Fitting a mixture model by expectation maximization to discover
From: Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology August, 1994, pp. 28-36. AAAI Press Fitting a mixture model by expectation maximization to discover
A SURVEY OF PLANTED MOTIF SEARCH ALGORITHMS
 A SURVEY OF PLANTED MOTIF SEARCH ALGORITHMS Adrian Kwok adriank@sfu.ca SFU ID #200136389 Literature Review CMPT 711 Professor Wiese February 23 rd, 2011 1. INTRODUCTION In Bioinformatics, discovering patterns
A SURVEY OF PLANTED MOTIF SEARCH ALGORITHMS Adrian Kwok adriank@sfu.ca SFU ID #200136389 Literature Review CMPT 711 Professor Wiese February 23 rd, 2011 1. INTRODUCTION In Bioinformatics, discovering patterns
1 Methods for Posterior Simulation
 1 Methods for Posterior Simulation Let p(θ y) be the posterior. simulation. Koop presents four methods for (posterior) 1. Monte Carlo integration: draw from p(θ y). 2. Gibbs sampler: sequentially drawing
1 Methods for Posterior Simulation Let p(θ y) be the posterior. simulation. Koop presents four methods for (posterior) 1. Monte Carlo integration: draw from p(θ y). 2. Gibbs sampler: sequentially drawing
CS Introduction to Data Mining Instructor: Abdullah Mueen
 CS 591.03 Introduction to Data Mining Instructor: Abdullah Mueen LECTURE 8: ADVANCED CLUSTERING (FUZZY AND CO -CLUSTERING) Review: Basic Cluster Analysis Methods (Chap. 10) Cluster Analysis: Basic Concepts
CS 591.03 Introduction to Data Mining Instructor: Abdullah Mueen LECTURE 8: ADVANCED CLUSTERING (FUZZY AND CO -CLUSTERING) Review: Basic Cluster Analysis Methods (Chap. 10) Cluster Analysis: Basic Concepts
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Sequence alignment algorithms
 Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
 Hindawi Publishing Corporation Mathematical Problems in Engineering Volume 2012, Article ID 490649, 9 pages doi:10.1155/2012/490649 Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
Hindawi Publishing Corporation Mathematical Problems in Engineering Volume 2012, Article ID 490649, 9 pages doi:10.1155/2012/490649 Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
Phylogenetics on CUDA (Parallel) Architectures Bradly Alicea
 Architectures Bradly Alicea") Descent w/modification Descent w/modification Descent w/modification Descent w/modification CPU Descent w/modification Descent w/modification Phylogenetics on CUDA (Parallel) Architectures Bradly Alicea
Descent w/modification Descent w/modification Descent w/modification Descent w/modification CPU Descent w/modification Descent w/modification Phylogenetics on CUDA (Parallel) Architectures Bradly Alicea
Semi-Supervised Clustering with Partial Background Information
 Semi-Supervised Clustering with Partial Background Information Jing Gao Pang-Ning Tan Haibin Cheng Abstract Incorporating background knowledge into unsupervised clustering algorithms has been the subject
Semi-Supervised Clustering with Partial Background Information Jing Gao Pang-Ning Tan Haibin Cheng Abstract Incorporating background knowledge into unsupervised clustering algorithms has been the subject
Using Hidden Markov Models for Multiple Sequence Alignments Lab #3 Chem 389 Kelly M. Thayer
 Página 1 de 10 Using Hidden Markov Models for Multiple Sequence Alignments Lab #3 Chem 389 Kelly M. Thayer Resources: Bioinformatics, David Mount Ch. 4 Multiple Sequence Alignments http://www.netid.com/index.html
Página 1 de 10 Using Hidden Markov Models for Multiple Sequence Alignments Lab #3 Chem 389 Kelly M. Thayer Resources: Bioinformatics, David Mount Ch. 4 Multiple Sequence Alignments http://www.netid.com/index.html
Clustering. CE-717: Machine Learning Sharif University of Technology Spring Soleymani
 Clustering CE-717: Machine Learning Sharif University of Technology Spring 2016 Soleymani Outline Clustering Definition Clustering main approaches Partitional (flat) Hierarchical Clustering validation
Clustering CE-717: Machine Learning Sharif University of Technology Spring 2016 Soleymani Outline Clustering Definition Clustering main approaches Partitional (flat) Hierarchical Clustering validation
CS281 Section 9: Graph Models and Practical MCMC
 CS281 Section 9: Graph Models and Practical MCMC Scott Linderman November 11, 213 Now that we have a few MCMC inference algorithms in our toolbox, let s try them out on some random graph models. Graphs
CS281 Section 9: Graph Models and Practical MCMC Scott Linderman November 11, 213 Now that we have a few MCMC inference algorithms in our toolbox, let s try them out on some random graph models. Graphs
Manual, ver. 03/01/2008, for PhylCRM_1.1 and the associated program PhylCRM_preprocess_1.1 utilized in the paper:
 Manual, ver. 03/01/2008, for PhylCRM_1.1 and the associated program PhylCRM_preprocess_1.1 utilized in the paper: Jason B. Warner 1,6, Anthony A. Philippakis 1,3,4,6, Savina A. Jaeger 1,6, Fangxue Sherry
Manual, ver. 03/01/2008, for PhylCRM_1.1 and the associated program PhylCRM_preprocess_1.1 utilized in the paper: Jason B. Warner 1,6, Anthony A. Philippakis 1,3,4,6, Savina A. Jaeger 1,6, Fangxue Sherry
Probabilistic Graphical Models
 Probabilistic Graphical Models Lecture 17 EM CS/CNS/EE 155 Andreas Krause Announcements Project poster session on Thursday Dec 3, 4-6pm in Annenberg 2 nd floor atrium! Easels, poster boards and cookies
Probabilistic Graphical Models Lecture 17 EM CS/CNS/EE 155 Andreas Krause Announcements Project poster session on Thursday Dec 3, 4-6pm in Annenberg 2 nd floor atrium! Easels, poster boards and cookies
Fall 09, Homework 5
 5-38 Fall 09, Homework 5 Due: Wednesday, November 8th, beginning of the class You can work in a group of up to two people. This group does not need to be the same group as for the other homeworks. You
5-38 Fall 09, Homework 5 Due: Wednesday, November 8th, beginning of the class You can work in a group of up to two people. This group does not need to be the same group as for the other homeworks. You
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Mixture Models and the EM Algorithm
 Mixture Models and the EM Algorithm Padhraic Smyth, Department of Computer Science University of California, Irvine c 2017 1 Finite Mixture Models Say we have a data set D = {x 1,..., x N } where x i is
Mixture Models and the EM Algorithm Padhraic Smyth, Department of Computer Science University of California, Irvine c 2017 1 Finite Mixture Models Say we have a data set D = {x 1,..., x N } where x i is
An Ultrafast Scalable Many-core Motif Discovery Algorithm for Multiple GPUs
 2011 IEEE International Parallel & Distributed Processing Symposium An Ultrafast Scalable Many-core Motif Discovery Algorithm for Multiple GPUs Yongchao Liu, Bertil Schmidt, Douglas L. Maskell School of
2011 IEEE International Parallel & Distributed Processing Symposium An Ultrafast Scalable Many-core Motif Discovery Algorithm for Multiple GPUs Yongchao Liu, Bertil Schmidt, Douglas L. Maskell School of
Using Real-valued Meta Classifiers to Integrate and Contextualize Binding Site Predictions
 Using Real-valued Meta Classifiers to Integrate and Contextualize Binding Site Predictions Offer Sharabi, Yi Sun, Mark Robinson, Rod Adams, Rene te Boekhorst, Alistair G. Rust, Neil Davey University of
Using Real-valued Meta Classifiers to Integrate and Contextualize Binding Site Predictions Offer Sharabi, Yi Sun, Mark Robinson, Rod Adams, Rene te Boekhorst, Alistair G. Rust, Neil Davey University of
ADAPTIVE METROPOLIS-HASTINGS SAMPLING, OR MONTE CARLO KERNEL ESTIMATION
 ADAPTIVE METROPOLIS-HASTINGS SAMPLING, OR MONTE CARLO KERNEL ESTIMATION CHRISTOPHER A. SIMS Abstract. A new algorithm for sampling from an arbitrary pdf. 1. Introduction Consider the standard problem of
ADAPTIVE METROPOLIS-HASTINGS SAMPLING, OR MONTE CARLO KERNEL ESTIMATION CHRISTOPHER A. SIMS Abstract. A new algorithm for sampling from an arbitrary pdf. 1. Introduction Consider the standard problem of
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
CSE 549: Computational Biology
 CSE 549: Computational Biology Phylogenomics 1 slides marked with * by Carl Kingsford Tree of Life 2 * H5N1 Influenza Strains Salzberg, Kingsford, et al., 2007 3 * H5N1 Influenza Strains The 2007 outbreak
CSE 549: Computational Biology Phylogenomics 1 slides marked with * by Carl Kingsford Tree of Life 2 * H5N1 Influenza Strains Salzberg, Kingsford, et al., 2007 3 * H5N1 Influenza Strains The 2007 outbreak
Theoretical Concepts of Machine Learning
 Theoretical Concepts of Machine Learning Part 2 Institute of Bioinformatics Johannes Kepler University, Linz, Austria Outline 1 Introduction 2 Generalization Error 3 Maximum Likelihood 4 Noise Models 5
Theoretical Concepts of Machine Learning Part 2 Institute of Bioinformatics Johannes Kepler University, Linz, Austria Outline 1 Introduction 2 Generalization Error 3 Maximum Likelihood 4 Noise Models 5
Lecture 8: Linkage algorithms and web search
 Lecture 8: Linkage algorithms and web search Information Retrieval Computer Science Tripos Part II Ronan Cummins 1 Natural Language and Information Processing (NLIP) Group ronan.cummins@cl.cam.ac.uk 2017
Lecture 8: Linkage algorithms and web search Information Retrieval Computer Science Tripos Part II Ronan Cummins 1 Natural Language and Information Processing (NLIP) Group ronan.cummins@cl.cam.ac.uk 2017
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment
 Multiple Sequence Alignment") CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/30/07 CAP5510 1 BLAST & FASTA FASTA [Lipman, Pearson 85, 88]
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/30/07 CAP5510 1 BLAST & FASTA FASTA [Lipman, Pearson 85, 88]
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment
 Multiple Sequence Alignment") GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
10701 Machine Learning. Clustering
 171 Machine Learning Clustering What is Clustering? Organizing data into clusters such that there is high intra-cluster similarity low inter-cluster similarity Informally, finding natural groupings among
171 Machine Learning Clustering What is Clustering? Organizing data into clusters such that there is high intra-cluster similarity low inter-cluster similarity Informally, finding natural groupings among
BCRANK: predicting binding site consensus from ranked DNA sequences
 BCRANK: predicting binding site consensus from ranked DNA sequences Adam Ameur October 30, 2017 1 Introduction This document describes the BCRANK R package. BCRANK[1] is a method that takes a ranked list
BCRANK: predicting binding site consensus from ranked DNA sequences Adam Ameur October 30, 2017 1 Introduction This document describes the BCRANK R package. BCRANK[1] is a method that takes a ranked list
Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh
 Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh Overlap detection: Semi-Global Alignment An overlap of two sequences is considered an
Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh Overlap detection: Semi-Global Alignment An overlap of two sequences is considered an
Spatial Patterns Point Pattern Analysis Geographic Patterns in Areal Data
 Spatial Patterns We will examine methods that are used to analyze patterns in two sorts of spatial data: Point Pattern Analysis - These methods concern themselves with the location information associated
Spatial Patterns We will examine methods that are used to analyze patterns in two sorts of spatial data: Point Pattern Analysis - These methods concern themselves with the location information associated
An Improved Algebraic Attack on Hamsi-256
 An Improved Algebraic Attack on Hamsi-256 Itai Dinur and Adi Shamir Computer Science department The Weizmann Institute Rehovot 76100, Israel Abstract. Hamsi is one of the 14 second-stage candidates in
An Improved Algebraic Attack on Hamsi-256 Itai Dinur and Adi Shamir Computer Science department The Weizmann Institute Rehovot 76100, Israel Abstract. Hamsi is one of the 14 second-stage candidates in
From Smith-Waterman to BLAST
 From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
version 1.0 User guide Ralf Eggeling, Ivo Grosse, Jan Grau
 version 1.0 User guide Ralf Eggeling, Ivo Grosse, Jan Grau If you use InMoDe, please cite: R. Eggeling, I. Grosse, and J. Grau. InMoDe: tools for learning and visualizing intra-motif dependencies of DNA
version 1.0 User guide Ralf Eggeling, Ivo Grosse, Jan Grau If you use InMoDe, please cite: R. Eggeling, I. Grosse, and J. Grau. InMoDe: tools for learning and visualizing intra-motif dependencies of DNA
CS224W Project Write-up Static Crawling on Social Graph Chantat Eksombatchai Norases Vesdapunt Phumchanit Watanaprakornkul
 1 CS224W Project Write-up Static Crawling on Social Graph Chantat Eksombatchai Norases Vesdapunt Phumchanit Watanaprakornkul Introduction Our problem is crawling a static social graph (snapshot). Given
1 CS224W Project Write-up Static Crawling on Social Graph Chantat Eksombatchai Norases Vesdapunt Phumchanit Watanaprakornkul Introduction Our problem is crawling a static social graph (snapshot). Given
Clustering Relational Data using the Infinite Relational Model
 Clustering Relational Data using the Infinite Relational Model Ana Daglis Supervised by: Matthew Ludkin September 4, 2015 Ana Daglis Clustering Data using the Infinite Relational Model September 4, 2015
Clustering Relational Data using the Infinite Relational Model Ana Daglis Supervised by: Matthew Ludkin September 4, 2015 Ana Daglis Clustering Data using the Infinite Relational Model September 4, 2015
ICB Fall G4120: Introduction to Computational Biology. Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology
 ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form.
 1 2 Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form. But note: invoking the algorithm is trivial. Using it correctly and interpreting the results, perhaps not so much.
1 2 Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form. But note: invoking the algorithm is trivial. Using it correctly and interpreting the results, perhaps not so much.
Protein Sequence Classification Using Probabilistic Motifs and Neural Networks
 Protein Sequence Classification Using Probabilistic Motifs and Neural Networks Konstantinos Blekas, Dimitrios I. Fotiadis, and Aristidis Likas Department of Computer Science, University of Ioannina, 45110
Protein Sequence Classification Using Probabilistic Motifs and Neural Networks Konstantinos Blekas, Dimitrios I. Fotiadis, and Aristidis Likas Department of Computer Science, University of Ioannina, 45110
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS 442 Computer vision. Fitting methods
 EECS 442 Computer vision Fitting methods - Problem formulation - Least square methods - RANSAC - Hough transforms - Multi-model fitting - Fitting helps matching! Reading: [HZ] Chapters: 4, 11 [FP] Chapters:
EECS 442 Computer vision Fitting methods - Problem formulation - Least square methods - RANSAC - Hough transforms - Multi-model fitting - Fitting helps matching! Reading: [HZ] Chapters: 4, 11 [FP] Chapters:
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
Treewidth and graph minors
 Treewidth and graph minors Lectures 9 and 10, December 29, 2011, January 5, 2012 We shall touch upon the theory of Graph Minors by Robertson and Seymour. This theory gives a very general condition under
Treewidth and graph minors Lectures 9 and 10, December 29, 2011, January 5, 2012 We shall touch upon the theory of Graph Minors by Robertson and Seymour. This theory gives a very general condition under
Assignment 2. Unsupervised & Probabilistic Learning. Maneesh Sahani Due: Monday Nov 5, 2018
 Assignment 2 Unsupervised & Probabilistic Learning Maneesh Sahani Due: Monday Nov 5, 2018 Note: Assignments are due at 11:00 AM (the start of lecture) on the date above. he usual College late assignments
Assignment 2 Unsupervised & Probabilistic Learning Maneesh Sahani Due: Monday Nov 5, 2018 Note: Assignments are due at 11:00 AM (the start of lecture) on the date above. he usual College late assignments
Collective Entity Resolution in Relational Data
 Collective Entity Resolution in Relational Data I. Bhattacharya, L. Getoor University of Maryland Presented by: Srikar Pyda, Brett Walenz CS590.01 - Duke University Parts of this presentation from: http://www.norc.org/pdfs/may%202011%20personal%20validation%20and%20entity%20resolution%20conference/getoorcollectiveentityresolution
Collective Entity Resolution in Relational Data I. Bhattacharya, L. Getoor University of Maryland Presented by: Srikar Pyda, Brett Walenz CS590.01 - Duke University Parts of this presentation from: http://www.norc.org/pdfs/may%202011%20personal%20validation%20and%20entity%20resolution%20conference/getoorcollectiveentityresolution
Identifying network modules
 Network biology minicourse (part 3) Algorithmic challenges in genomics Identifying network modules Roded Sharan School of Computer Science, Tel Aviv University Gene/Protein Modules A module is a set of
Network biology minicourse (part 3) Algorithmic challenges in genomics Identifying network modules Roded Sharan School of Computer Science, Tel Aviv University Gene/Protein Modules A module is a set of
Algorithms for Integer Programming
 Algorithms for Integer Programming Laura Galli November 9, 2016 Unlike linear programming problems, integer programming problems are very difficult to solve. In fact, no efficient general algorithm is
Algorithms for Integer Programming Laura Galli November 9, 2016 Unlike linear programming problems, integer programming problems are very difficult to solve. In fact, no efficient general algorithm is
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
15-780: Graduate Artificial Intelligence. Computational biology: Sequence alignment and profile HMMs
 5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
Parsimony-Based Approaches to Inferring Phylogenetic Trees
 Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Fitting. Instructor: Jason Corso (jjcorso)! web.eecs.umich.edu/~jjcorso/t/598f14!! EECS Fall 2014! Foundations of Computer Vision!
! web.eecs.umich.edu/~jjcorso/t/598f14!! EECS Fall 2014! Foundations of Computer Vision!") Fitting EECS 598-08 Fall 2014! Foundations of Computer Vision!! Instructor: Jason Corso (jjcorso)! web.eecs.umich.edu/~jjcorso/t/598f14!! Readings: FP 10; SZ 4.3, 5.1! Date: 10/8/14!! Materials on these
Fitting EECS 598-08 Fall 2014! Foundations of Computer Vision!! Instructor: Jason Corso (jjcorso)! web.eecs.umich.edu/~jjcorso/t/598f14!! Readings: FP 10; SZ 4.3, 5.1! Date: 10/8/14!! Materials on these
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Midterm Examination CS540-2: Introduction to Artificial Intelligence
 Midterm Examination CS540-2: Introduction to Artificial Intelligence March 15, 2018 LAST NAME: FIRST NAME: Problem Score Max Score 1 12 2 13 3 9 4 11 5 8 6 13 7 9 8 16 9 9 Total 100 Question 1. [12] Search
Midterm Examination CS540-2: Introduction to Artificial Intelligence March 15, 2018 LAST NAME: FIRST NAME: Problem Score Max Score 1 12 2 13 3 9 4 11 5 8 6 13 7 9 8 16 9 9 Total 100 Question 1. [12] Search
University of Wisconsin-Madison Spring 2018 BMI/CS 776: Advanced Bioinformatics Homework #2
 Assignment goals Use mutual information to reconstruct gene expression networks Evaluate classifier predictions Examine Gibbs sampling for a Markov random field Control for multiple hypothesis testing
Assignment goals Use mutual information to reconstruct gene expression networks Evaluate classifier predictions Examine Gibbs sampling for a Markov random field Control for multiple hypothesis testing
Deep Reinforcement Learning
 Deep Reinforcement Learning 1 Outline 1. Overview of Reinforcement Learning 2. Policy Search 3. Policy Gradient and Gradient Estimators 4. Q-prop: Sample Efficient Policy Gradient and an Off-policy Critic
Deep Reinforcement Learning 1 Outline 1. Overview of Reinforcement Learning 2. Policy Search 3. Policy Gradient and Gradient Estimators 4. Q-prop: Sample Efficient Policy Gradient and an Off-policy Critic
Clustering gene expression data
 Clustering gene expression data 1 How Gene Expression Data Looks Entries of the Raw Data matrix: Ratio values Absolute values Row = gene s expression pattern Column = experiment/condition s profile genes
Clustering gene expression data 1 How Gene Expression Data Looks Entries of the Raw Data matrix: Ratio values Absolute values Row = gene s expression pattern Column = experiment/condition s profile genes
Recent Research Results. Evolutionary Trees Distance Methods
 Recent Research Results Evolutionary Trees Distance Methods Indo-European Languages After Tandy Warnow What is the purpose? Understand evolutionary history (relationship between species). Uderstand how
Recent Research Results Evolutionary Trees Distance Methods Indo-European Languages After Tandy Warnow What is the purpose? Understand evolutionary history (relationship between species). Uderstand how
GPU-MEME: Using Graphics Hardware to Accelerate Motif Finding in DNA Sequences
 GPU-MEME: Using Graphics Hardware to Accelerate Motif Finding in DNA Sequences Chen Chen, Bertil Schmidt, Liu Weiguo, and Wolfgang Müller-Wittig School of Computer Engineering, Nanyang Technological University,
GPU-MEME: Using Graphics Hardware to Accelerate Motif Finding in DNA Sequences Chen Chen, Bertil Schmidt, Liu Weiguo, and Wolfgang Müller-Wittig School of Computer Engineering, Nanyang Technological University,
Clustering Lecture 5: Mixture Model
 Clustering Lecture 5: Mixture Model Jing Gao SUNY Buffalo 1 Outline Basics Motivation, definition, evaluation Methods Partitional Hierarchical Density-based Mixture model Spectral methods Advanced topics
Clustering Lecture 5: Mixture Model Jing Gao SUNY Buffalo 1 Outline Basics Motivation, definition, evaluation Methods Partitional Hierarchical Density-based Mixture model Spectral methods Advanced topics
1 More configuration model
 1 More configuration model In the last lecture, we explored the definition of the configuration model, a simple method for drawing networks from the ensemble, and derived some of its mathematical properties.
1 More configuration model In the last lecture, we explored the definition of the configuration model, a simple method for drawing networks from the ensemble, and derived some of its mathematical properties.
Breaking Grain-128 with Dynamic Cube Attacks
 Breaking Grain-128 with Dynamic Cube Attacks Itai Dinur and Adi Shamir Computer Science department The Weizmann Institute Rehovot 76100, Israel Abstract. We present a new variant of cube attacks called
Breaking Grain-128 with Dynamic Cube Attacks Itai Dinur and Adi Shamir Computer Science department The Weizmann Institute Rehovot 76100, Israel Abstract. We present a new variant of cube attacks called
Richard Feynman, Lectures on Computation
 Chapter 8 Sorting and Sequencing If you keep proving stuff that others have done, getting confidence, increasing the complexities of your solutions for the fun of it then one day you ll turn around and
Chapter 8 Sorting and Sequencing If you keep proving stuff that others have done, getting confidence, increasing the complexities of your solutions for the fun of it then one day you ll turn around and
Final Exam. Introduction to Artificial Intelligence. CS 188 Spring 2010 INSTRUCTIONS. You have 3 hours.
 CS 188 Spring 2010 Introduction to Artificial Intelligence Final Exam INSTRUCTIONS You have 3 hours. The exam is closed book, closed notes except a two-page crib sheet. Please use non-programmable calculators
CS 188 Spring 2010 Introduction to Artificial Intelligence Final Exam INSTRUCTIONS You have 3 hours. The exam is closed book, closed notes except a two-page crib sheet. Please use non-programmable calculators
CisGenome User s Manual
 CisGenome User s Manual 1. Overview 1.1 Basic Framework of CisGenome 1.2 Installation 1.3 Summary of All Functions 1.4 A Quick Start Analysis of a ChIP-chip Experiment 2. Genomics Toolbox I Establishing
CisGenome User s Manual 1. Overview 1.1 Basic Framework of CisGenome 1.2 Installation 1.3 Summary of All Functions 1.4 A Quick Start Analysis of a ChIP-chip Experiment 2. Genomics Toolbox I Establishing
Manual, ver. 03/01/2008, for Lever_1.1, and the associated programs PhylCRM_preprocess_1.1 and Lever_statistics_1.1 utilized in the paper:
 Manual, ver. 03/01/2008, for Lever_1.1, and the associated programs PhylCRM_preprocess_1.1 and Lever_statistics_1.1 utilized in the paper: Jason B. Warner 1,6, Anthony A. Philippakis 1,3,4,6, Savina A.
Manual, ver. 03/01/2008, for Lever_1.1, and the associated programs PhylCRM_preprocess_1.1 and Lever_statistics_1.1 utilized in the paper: Jason B. Warner 1,6, Anthony A. Philippakis 1,3,4,6, Savina A.
Dynamic Programming: Sequence alignment. CS 466 Saurabh Sinha
 Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Locality-sensitive hashing and biological network alignment
 Locality-sensitive hashing and biological network alignment Laura LeGault - University of Wisconsin, Madison 12 May 2008 Abstract Large biological networks contain much information about the functionality
Locality-sensitive hashing and biological network alignment Laura LeGault - University of Wisconsin, Madison 12 May 2008 Abstract Large biological networks contain much information about the functionality