3.4 Multiple sequence alignment
|
|
|
- Arron Daniel
- 5 years ago
- Views:
Transcription
1 3.4 Multiple sequence alignment Why produce a multiple sequence alignment? Using more than two sequences results in a more convincing alignment by revealing conserved regions in ALL of the sequences Aligned sequences also allow for conservative substitutions and gaps among the a conserved family of sequences Conserved regions/domains are likely to represent regions that are essential for structure and function - core of proteins A multiple sequence alignment is a starting point for an evolutionary (phylogenetic) analysis
2 Types of multiple sequence alignment Global alignment in which entire sequences are aligned at the same time using extension of dynamic programming Local alignment in which conserved local regions are found by statistical methods Local alignment derived by removing stretches of global alignment
3 Example of local msa Seq. 1 Seq. 3 Seq. 5 Seq. 2 Seq. 4 OBJECT: to find an identifiable common, usually longest, pattern with some degree of variability. No gaps are shown in this example but they can be accomodated. AGGCTT AAGCTA AGACTT TAACTA AGACTT
4 The flowchart of multisequence alignment
5 Challenges Finding an optimal alignment of more than two sequences that includes matches, mismatches, and gaps, and that takes into account the degree of variation in all of the sequences at the same time poses a very difficult challenge. A second computational challenge is identifying a reasonable method of obtaining a cumulative score for the substitutions in the column of an msa.
6 Problems: Multiple sequence alignment is computational complex Suppose one tries to align three sequences by extending the method of aligning 2 sequences to a 3 dimensional scoring matrix. Sequence 1 Sequence 3 W Y Time and space needed is length of seq. raised to power of no. of sequences Can do for three sequences but not more than three Sequence 2 W? Optimal score in 3 dimensions
7 Alignment of three sequences by dynamic programming For three protein sequences each 300 amino acids in length and excluding gaps, the number of comparisons to be made by dynamic programming is equal to = , whereas only = is required for two sequences of this length. (The number of steps and memory required for N M-amino-acid sequences: M N ) Carrillo and Lipman (1988) found a way (the sum of pairs, SP method) to reduce the number of comparisons that must be made without compromising the attempt to find an optimal alignment. the MSA program,

8 Basic idea of MSA program: sum of pairs (SP) method
9 The SP model for scoring a msa A: phylogenetic tree B: star phylogeny
10 Thus, approximate methods are used, including: (1) a progressive global alignment of the sequences starting with an alignment of the most alike sequences and then building an alignment by adding more sequences; (2) iterative methods that make an initial alignment of groups of sequences and then revise the alignment to achieve a more reasonable result; (3) alignments based on locally conserved patterns found in the same order in the sequences; (4) use of statistical methods and probabilistic models of the sequences.
11 3.4.1 PROGRESSIVE METHODS OF GLOBAL MULTIPLE SEQUENCE ALIGNMENT Progressive alignment methods use the dynamic programming method to build a msa starting with the most related sequences and then progressively adding less-related sequences or groups of sequences to the initial alignment (Waterman and Perlwitz 1984; Feng and Doolittle 1987, 1996; Thompson et al. 1994a; Higgins et al. 1996). Relationships among the sequences are modeled by an evolutionary tree in which the outer branches or leaves are the sequences
12 The tree is based on pair-wise comparisons of the sequences using one of the phylogenetic methods Global multiple sequence alignment The methods are used by CLUSTALW and the Genetics Computer Group program PILEUP.

13 CLUSTALW CLUSTAL has been around for more than 10 years, and the authors have done much to support and improve the program (Higgins and Sharp 1988; Thompson et al. 1994a; Higgins et al. 1996). CLUSTALW is a more recent version of CLUSTAL with the W standing for weighting to represent the ability of the program to provide weights to the sequence and program parameters, and CLUSTALX provides a graphic interface

14 Baxenavis and Oullette, 2001
15 The steps include: (1) Perform pair-wise alignments of all of the sequences; (2) use the alignment scores to produce a phylogenetic tree (for an explanation of the neighbor-joining method that is used); and (3) align the sequences sequentially, guided by the phylogenetic relationships indicated by the tree. The initial alignments used to produce the guide tree may be obtained by a fast k-tuple or patternfinding approach similar to FASTA that is useful for many sequences, or a slower, full dynamic programming method may be used. An enhanced dynamic programming alignment algorithm (Myers and Miller 1988) is used to obtain optimal alignment scores.
16 Weighting scheme used by CLUSTALW
17 The scoring of gaps in a msa has to be performed in a different manner from scoring gaps in a pair-wise alignment. CLUSTALW calculates gaps in a novel way designed to place them between conserved domains. Such as Pascarella and Argos (1992) aligned sequences of structurally related proteins, the gaps were preferentially found between secondary structural elements. These authors also prepared a table of the observed frequency of gaps next to each amino acid in these regions. CLUSTALW uses the information in this table.
18 CLUSTALW also attempts to locate what may be the corresponding domains by appropriate gap placement in the msa. GAP SEPARATION DISTANCE tries to decrease the chances of gaps being too close to each other. Gaps that are less than this distance apart are penalised more than other gaps. END GAP SEPARATION treats end gaps just like internal gaps for the purposes of avoiding gaps that are too close (set by GAP SEPARATION DISTANCE above). If you turn this off, end gaps will be ignored for this purpose. This is useful when you wish to align fragments where the end gaps are not biologically meaningful.

19 Example of Sequence Alignment using Clustal W Asterisk represents identity : represents high similarity. represents low similarity
20 Features of Progressive Multiple Sequence Alignment handles the alignment of domains very well sequences must match closely enough to obtain global alignment but must not be too alike since alike sequences bias the multiple sequence alignment the first alignments between the most alike sequences determine the alignment with all of the other sequences
21 Advantages and disadvantages The major problem with the progressive alignment method described above is that errors in the initial alignments of the most closely related sequences are propagated to the msa. For the difficult task of aligning more distantly related sequences, using Bayesian methods such as hidden Markov models (HMMs) may be useful. For more closely relate sequences, CLUSTALW is designed to provide an adequate alignment of a large number of sequences and provide a very good indication of the domain structure of those sequences.
22 3.4.2 ITERATIVE METHODS OF MULTIPLE SEQUENCE ALIGNMENT Iterative methods is by repeatedly realigning subgroups of the sequences and then by aligning these subgroups into a global alignment of all of the sequences. The objective is to improve the overall alignment score, such as a sum of pairs score. Selection of these groups may be based on the ordering of the sequences on a phylogenetic tree predicted in a manner similar to that of progressive alignment, separation of one or two of the sequences from the rest, or a random selection of the groups.
23 Iterative MSA methods The idea is to start with a reasonable approximation to the optimal MSA (e.g. by using a progressive method) and then tweaking to improve it. Various optimization techniques have been tried (e.g. GAs and simulated annealing). Key is the scoring function for the whole MSA. Also, what steps to take that are likely to improve the score
24 Block based methods Another approach to iterative methods are to start with short local alignments (sometimes called blocks) and then to reduce the problem to aligning the regions between the blocks Divide and conquer is another common CS approach to exponential problems. How to find the blocks? DALIGN (local alignment methods) DCA (divide and conquer alignments) Tmsa (identify patterns and use them to define blocks).
25 Programs MultAlin (Corpet 1988), PRRP, DIALIGN SAGA (Notredame and Higgins,1996) : The genetic algorithm is a general type of machine-learning algorithm that has no direct relationship to biology and that was invented by computer scientists. Hidden Markov Models: SAM (Sequence Alignment and Modeling Software System) (Krogh et al. 1994; Hughey and Krogh 1996); HMMER (Eddy 1998)
26 3.4.3 Local alignment of multiple sequences Multiple sequence alignment using PSSMs, the E/M method Multiple sequence alignment using Gibbs and HMMs
27 Local alignment of multiple sequences! Object is to find if a set of sequences share one or more common patterns of same length! Pattern may be short or may include most of the sequences! Pattern may include substitutions! Pattern may or may not include gaps Rest of proteins do not align well Domain that aligns well Rest of proteins do not align well
28 Example of local msa Seq. 1 Seq. 3 Seq. 5 Seq. 2 Seq. 4 OBJECT: to find an identifiable common, usually longest, pattern with some degree of variability. No gaps are shown in this example but they can be accomodated. AGGCTT AAGCTA AGACTT TAACTA AGACTT
29 Starting from a global multiple sequence alignment! Already have global multiple sequence alignment Locate a well conserved region Extract that region from the alignment Model the local alignment " position-specific scoring matrix (PSSM) " sequence profile " profile hidden Markov model
30 Starting without a global multiple sequence alignment (2)! Do not already have global multiple sequence alignment! Find common patterns in sequences simple hash methods statistical methods " Expectation maximization " Gibbs sampling! Align the sequences by these patterns
31 Simple hash method for sequences that have not been aligned (used by FASTA) - Make a list of words of length n for each sequence - Make an index of these words for each sequence showing sequence position where word is found - Look for similar words present in all sequences - Word position provides the alignment - Can use several words to provide a longer alignment - Finding fixed (but sparse) patterns
32
33 Removing a local alignment from a global alignment to make a PSSM Alignment AGGCTT AAGCTA AGACTT TAACTA AGACTT Fraction of base in each column Column Sum I II III IV V VI A G C T Column Sum I II III IV V VI A G C T
34 Making a scoring matrix (cont d) Fraction divided by bdg. freq. Column Sum Bdg I II III IV V VI freq. A G C T sum Convert to log to base 2 A G C T A PSSM (POSITION SPECIFIC SCORING MATRIX)
35 Aligning the new scoring matrix with a new sequence provides a log odds alignment score New sequence... A G A C T A... Log odds matrix or PSSM A G C T Conclusion: Log odds score at this sequence position = 5.2 Odds score = = 37/1 in favor of not being a random match
36 Using the expectation maximization (E/M) method to make a PSSM Two steps: expectation step (1-7) and maximization step (8) 1. Make a random alignment of the sequences by picking a random position in each. 2. Choose an arbitrary pattern width. 3. Make a trial log odds scoring matrix. Sequences randomly aligned Trial PSSM
37 E/M method Slide the trial PSSM along each sequence 5. Measure odds of matching each sequence position /1 1/25 33/1 1/ Sum the odds scores e.g and then calculate the probability that trial PSSM matches each position e.g. 100/5000 (0.02) 0.04/ /5000 1/ Repeat for all of the sequences.
38 E/M method Update the table with the information found from scanning the sequences (maximization step). e.g. suppose that. - there are 100 A's in the first column of the original trial alignment - that a match to sequence 1 was found with a P = that there was an A in the sequence at position 1 A 100 A's P of match at this position in sequence = 0.1 Add 0.1 A's to count in column 1
39 E/M method 4 9. Update table using all information from scanning all of the sequences, make a new table, scan sequences again times (the expectation and maximization steps are repeated until the estimates of the base frequencies do not change) 10. End result: a PSSM table that represents one alignment position A G C T
40 How to Evaluate the Quality of a PSSM! How well does the PSSM find matches in a new sequences - this is equivalent to asking how much information is present in the PSSM?! Calculate the information content of the PSSM! Information content in the scoring matrix reduces uncertainty
41 Meaning of Uncertainty Uncertainty is the no. of questions that must be asked to identify the correct choice i.e. the correct base or amino acid in one location of a particular pattern. Uncertainty is reduced by the information in the scoring matrix Example: consider 64 cups in a row with an object hidden under one of them. The goal is to find the object with as few questions as possible. (Answer: uncertainty is six and is zero when the object is found) G A C T How much uncertainty is there for: column 1, column 2 and column 3? For DNA sequences, what is the maximum uncertainty there can be? What is the minimum uncertainty?
42 Information and Probability I=logN=-logP, P=1/N (Hartly, 1928) I = - Σp i log 2 (p i ) (Shannon, 1948)
43 Calculating Uncertainty and Information Content for PSSM Uncertainty (H): H = f G log 2 (f G ) + f A log 2 (f A ) +... In general, the average amount of uncertainty (H c ) in bits per symbol for column c of the PSSM is given by for entire PSSM H c = - Σp ic log 2 (p ic ) All i H = ΣH c All columns H is also known as the entropy of the PSSM position in information theory because the higher the value, the greater the uncertainty. Information content (IC): IC for column = 2 - H IC for whole PSSM = sum of columns ICs

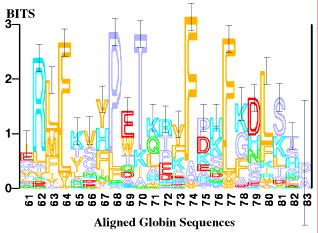
44 Sequence logos
45 Questions about quality of a PSSM! What is the highest information content we can have for a matrix with 6 columns representing DNA sequences?! How much sequence variation will be present in each column of the resulting matrix i.e. How much variation will be found in target sequences using this matrix?! Is information content the only factor to consider in a making a PSSM to represent a sequence alignment?! For proteins, what is the no. of questions that must be asked to identify the correct amino acid in one location of a particular pattern?
46 Local alignment by Gibbs Sampling! Gibbs sampling is another method for locating common patterns in a set of sequences! Has proven to be useful for promoter analysis, including self-complementary sequence patterns! Method like expectation maximization in starting with a random alignment of the sequences! Difference is that one sequence (randomly or deliberately chosen) is left out and that sequence is scanned for matches using the trial PSSM
47 Gibbs sampling (2) (1) The predictive update step: Start with random alignment but leave out one sequence. Left out sequence Left out sequence Trial PSSM from other sequences (2) Slide PSSM along left out sequence. Trial PSSM Probability distribution of pattern along sequence
48 Gibbs sampling (3)! Use probability distribution of pattern along sequence to choose a "trial, possible" location (as with E/M)! To see how this is done, imagine filling a bag with marbles marked with all possible locations of the pattern in the left out sequence! Weight the no. of each type according to the probability of a match at that site! Pick a random marble representing a weighted location 3 times as likely to pick a 12 as
49 Gibbs sampling (4)! Use weighted, but randomly chosen position as the location of the trial pattern in the left out sequence! Make a new PSSM of that sequence against a new set of random location in all other sequences but one! Repeat all steps with the new left out sequence! RESULT: after 100 or so iterations, the most alike patterns in the sequences are recruited into a best possible PSSM
50 Modeling Local Alignments with Gaps: Simple Profiles and Profile Hidden Markov Models EM and Gibbs are only used for alignments that involve patterns without gaps To include gaps, a profile is made Types - simple profile and a profile hidden Markov model* *HMMs may also be used to model multiple domains or global alignments
51 Sequence Profile - Definition! Simple Profile is a type of scoring matrix like a PSSM has extra rows (or columns) with gap penalties! Profile is aligned with a sequence using the dynamic programming algorithm! Produces an optimal alignment of the profile with the sequence! If high score found, then the alignment is significant! Not used as much as a profile HMM
52 Hypothetical profile showing log odds scores and gap penalties Column I II III IV V VI A G C T Gap Open Gap Ext Align with sequence using dynamic programming.
53 Markov Models! Have a series of events A, B, C, etc! The prob. of a particular current event is not influenced by previous ones! Example is sequence alignment - each aligned position is scored independently
54 Understanding Hidden Markov Models! Imagine that we survey the routes that are taken to drive from Tucson to NY and the hotels that people stay at.! Record trips for several 100 travellers including roads and hotels - training the model! Calculate chance of a new traveller going on one particular route and staying at one particular hotel. TUS-DEN(hotel B)-NY? TUS-DFW(hotel C)-NY?
55 Understanding Hidden Markov Models Prob. of a new traveler going to Salt Lake, AND staying at hotel A, AND going to Denver AND staying at hotel B, AND going direct to NY is? 0.1 TUS SLC A 0.7 B 0.1 C DEN A 0.3 B 0.3 C DFW A 0.2 B 0.3 C NY
56 What is hidden?! The probabilities of choosing routes are a Markov chain - assuming that each leg is independent! These probabilities are called transition probabilities! Cities are states in the model.! Associated with each state is a probability, that of staying at one particular hotel! These probabilities are hidden in that we have to ask what is the probability of A, etc.! In HMM s, the states emit a certain result (hotel letter, or sequence letter) with a given probability
57 HMMs are used for modeling sequence alignments Alignment: N. F L S N. F L S N K Y L T Q. W - T Represented by HMM which is "trained" on sequences Match states emit an amino acid with probability given by amino acid distribution in that state. Insert state emits any amino acid with equal frequency. Length of model corresponds to alignment length and columns to the columns on the alignment
58 Many profile HMMs are made representing protein alignments HMMs representing motifs, domains, etc. "Align each HMM model with a sequence to find most probable path through the model and the probability of a match of sequence to model. "High probability means a match to that profile. "Alignment algorithms used are similar in principle to dynamic programming
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment
 Multiple Sequence Alignment") CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
PROTEIN MULTIPLE ALIGNMENT MOTIVATION: BACKGROUND: Marina Sirota
 Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment
 Multiple Sequence Alignment") GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Lecture 5: Multiple sequence alignment
 Lecture 5: Multiple sequence alignment Introduction to Computational Biology Teresa Przytycka, PhD (with some additions by Martin Vingron) Why do we need multiple sequence alignment Pairwise sequence alignment
Lecture 5: Multiple sequence alignment Introduction to Computational Biology Teresa Przytycka, PhD (with some additions by Martin Vingron) Why do we need multiple sequence alignment Pairwise sequence alignment
Profiles and Multiple Alignments. COMP 571 Luay Nakhleh, Rice University
 Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Multiple Sequence Alignment. Mark Whitsitt - NCSA
 Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/30/07 CAP5510 1 BLAST & FASTA FASTA [Lipman, Pearson 85, 88]
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 1/30/07 CAP5510 1 BLAST & FASTA FASTA [Lipman, Pearson 85, 88]
Chapter 6. Multiple sequence alignment (week 10)
") Course organization Introduction ( Week 1,2) Part I: Algorithms for Sequence Analysis (Week 1-11) Chapter 1-3, Models and theories» Probability theory and Statistics (Week 3)» Algorithm complexity analysis
Course organization Introduction ( Week 1,2) Part I: Algorithms for Sequence Analysis (Week 1-11) Chapter 1-3, Models and theories» Probability theory and Statistics (Week 3)» Algorithm complexity analysis
Multiple Sequence Alignment Sum-of-Pairs and ClustalW. Ulf Leser
 Multiple Sequence Alignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence Alignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Multiple Sequence Alignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence Alignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Stephen Scott.
 1 / 33 sscott@cse.unl.edu 2 / 33 Start with a set of sequences In each column, residues are homolgous Residues occupy similar positions in 3D structure Residues diverge from a common ancestral residue
1 / 33 sscott@cse.unl.edu 2 / 33 Start with a set of sequences In each column, residues are homolgous Residues occupy similar positions in 3D structure Residues diverge from a common ancestral residue
Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods
 Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods Khaddouja Boujenfa, Nadia Essoussi, and Mohamed Limam International Science Index, Computer and Information Engineering waset.org/publication/482
Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods Khaddouja Boujenfa, Nadia Essoussi, and Mohamed Limam International Science Index, Computer and Information Engineering waset.org/publication/482
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
Multiple Sequence Alignment (MSA)
") I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT
 HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT - Swarbhanu Chatterjee. Hidden Markov models are a sophisticated and flexible statistical tool for the study of protein models. Using HMMs to analyze proteins
HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT - Swarbhanu Chatterjee. Hidden Markov models are a sophisticated and flexible statistical tool for the study of protein models. Using HMMs to analyze proteins
Chapter 8 Multiple sequence alignment. Chaochun Wei Spring 2018
 1896 1920 1987 2006 Chapter 8 Multiple sequence alignment Chaochun Wei Spring 2018 Contents 1. Reading materials 2. Multiple sequence alignment basic algorithms and tools how to improve multiple alignment
1896 1920 1987 2006 Chapter 8 Multiple sequence alignment Chaochun Wei Spring 2018 Contents 1. Reading materials 2. Multiple sequence alignment basic algorithms and tools how to improve multiple alignment
AlignMe Manual. Version 1.1. Rene Staritzbichler, Marcus Stamm, Kamil Khafizov and Lucy R. Forrest
 AlignMe Manual Version 1.1 Rene Staritzbichler, Marcus Stamm, Kamil Khafizov and Lucy R. Forrest Max Planck Institute of Biophysics Frankfurt am Main 60438 Germany 1) Introduction...3 2) Using AlignMe
AlignMe Manual Version 1.1 Rene Staritzbichler, Marcus Stamm, Kamil Khafizov and Lucy R. Forrest Max Planck Institute of Biophysics Frankfurt am Main 60438 Germany 1) Introduction...3 2) Using AlignMe
Jyoti Lakhani 1, Ajay Khunteta 2, Dharmesh Harwani *3 1 Poornima University, Jaipur & Maharaja Ganga Singh University, Bikaner, Rajasthan, India
 International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
MULTIPLE SEQUENCE ALIGNMENT
 MULTIPLE SEQUENCE ALIGNMENT Multiple Alignment versus Pairwise Alignment Up until now we have only tried to align two sequences. What about more than two? A faint similarity between two sequences becomes
MULTIPLE SEQUENCE ALIGNMENT Multiple Alignment versus Pairwise Alignment Up until now we have only tried to align two sequences. What about more than two? A faint similarity between two sequences becomes
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
Multiple Sequence Alignment II
 Multiple Sequence Alignment II Lectures 20 Dec 5, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline
Multiple Sequence Alignment II Lectures 20 Dec 5, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline
Multiple sequence alignment. November 20, 2018
 Multiple sequence alignment November 20, 2018 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one
Multiple sequence alignment November 20, 2018 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one
Lecture 5: Markov models
 Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS
 MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS By XU ZHANG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE
MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS By XU ZHANG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE
Genome 559: Introduction to Statistical and Computational Genomics. Lecture15a Multiple Sequence Alignment Larry Ruzzo
 Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Multiple Sequence Alignment: Multidimensional. Biological Motivation
 Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004
 CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004 Lecture #4: 8 April 2004 Topics: Sequence Similarity Scribe: Sonil Mukherjee 1 Introduction
CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004 Lecture #4: 8 April 2004 Topics: Sequence Similarity Scribe: Sonil Mukherjee 1 Introduction
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment
 Scoring Multiple Alignments Entropy Sum of Pairs Alignment") Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment Partial Order Alignment (POA) A-Bruijin (ABA) Approach
Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment Partial Order Alignment (POA) A-Bruijin (ABA) Approach
INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS
 INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS MUHANNAD A. ABU-HASHEM, NUR'AINI ABDUL RASHID, ROSNI ABDULLAH, AWSAN A. HASAN AND ATHEER A. ABDULRAZZAQ School of Computer Sciences, Universiti
INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS MUHANNAD A. ABU-HASHEM, NUR'AINI ABDUL RASHID, ROSNI ABDULLAH, AWSAN A. HASAN AND ATHEER A. ABDULRAZZAQ School of Computer Sciences, Universiti
In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace.
 5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Brief review from last class
 Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
A New Approach For Tree Alignment Based on Local Re-Optimization
 A New Approach For Tree Alignment Based on Local Re-Optimization Feng Yue and Jijun Tang Department of Computer Science and Engineering University of South Carolina Columbia, SC 29063, USA yuef, jtang
A New Approach For Tree Alignment Based on Local Re-Optimization Feng Yue and Jijun Tang Department of Computer Science and Engineering University of South Carolina Columbia, SC 29063, USA yuef, jtang
Comparison and Evaluation of Multiple Sequence Alignment Tools In Bininformatics
 IJCSNS International Journal of Computer Science and Network Security, VOL.9 No.7, July 2009 51 Comparison and Evaluation of Multiple Sequence Alignment Tools In Bininformatics Asieh Sedaghatinia, Dr Rodziah
IJCSNS International Journal of Computer Science and Network Security, VOL.9 No.7, July 2009 51 Comparison and Evaluation of Multiple Sequence Alignment Tools In Bininformatics Asieh Sedaghatinia, Dr Rodziah
JET 2 User Manual 1 INSTALLATION 2 EXECUTION AND FUNCTIONALITIES. 1.1 Download. 1.2 System requirements. 1.3 How to install JET 2
 JET 2 User Manual 1 INSTALLATION 1.1 Download The JET 2 package is available at www.lcqb.upmc.fr/jet2. 1.2 System requirements JET 2 runs on Linux or Mac OS X. The program requires some external tools
JET 2 User Manual 1 INSTALLATION 1.1 Download The JET 2 package is available at www.lcqb.upmc.fr/jet2. 1.2 System requirements JET 2 runs on Linux or Mac OS X. The program requires some external tools
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec2 Choosing the Right Sequences Choosing the Right Sequences Before you build your alignment,
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec2 Choosing the Right Sequences Choosing the Right Sequences Before you build your alignment,
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Phylogenetics. Introduction to Bioinformatics Dortmund, Lectures: Sven Rahmann. Exercises: Udo Feldkamp, Michael Wurst
 Phylogenetics Introduction to Bioinformatics Dortmund, 16.-20.07.2007 Lectures: Sven Rahmann Exercises: Udo Feldkamp, Michael Wurst 1 Phylogenetics phylum = tree phylogenetics: reconstruction of evolutionary
Phylogenetics Introduction to Bioinformatics Dortmund, 16.-20.07.2007 Lectures: Sven Rahmann Exercises: Udo Feldkamp, Michael Wurst 1 Phylogenetics phylum = tree phylogenetics: reconstruction of evolutionary
Bioinformatics explained: Smith-Waterman
 Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
PoMSA: An Efficient and Precise Position-based Multiple Sequence Alignment Technique
 PoMSA: An Efficient and Precise Position-based Multiple Sequence Alignment Technique Sara Shehab 1,a, Sameh Shohdy 1,b, Arabi E. Keshk 1,c 1 Department of Computer Science, Faculty of Computers and Information,
PoMSA: An Efficient and Precise Position-based Multiple Sequence Alignment Technique Sara Shehab 1,a, Sameh Shohdy 1,b, Arabi E. Keshk 1,c 1 Department of Computer Science, Faculty of Computers and Information,
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 13 G R A T I V. Iterative homology searching,
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón
 trimal: a tool for automated alignment trimming in large-scale phylogenetics analyses Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón Version 1.2b Index of contents 1. General features
trimal: a tool for automated alignment trimming in large-scale phylogenetics analyses Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón Version 1.2b Index of contents 1. General features
Machine Learning. Computational biology: Sequence alignment and profile HMMs
 10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
Parsimony-Based Approaches to Inferring Phylogenetic Trees
 Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Multiple Sequence Alignment Sum-of-Pairs and ClustalW. Ulf Leser
 Multiple Sequence lignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence lignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Multiple Sequence lignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence lignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
From [4] D. Gusfield. Algorithms on Strings, Trees and Sequences. Cambridge University Press, New York, 1997.
![From [4] D. Gusfield. Algorithms on Strings, Trees and Sequences. Cambridge University Press, New York, 1997.](/thumbs/85/92404365.jpg "From [4] D. Gusfield. Algorithms on Strings, Trees and Sequences. Cambridge University Press, New York, 1997.") Multiple Alignment From [4] D. Gusfield. Algorithms on Strings, Trees and Sequences. Cambridge University Press, New York, 1997. 1 Multiple Sequence Alignment Introduction Very active research area Very
Multiple Alignment From [4] D. Gusfield. Algorithms on Strings, Trees and Sequences. Cambridge University Press, New York, 1997. 1 Multiple Sequence Alignment Introduction Very active research area Very
BLAST - Basic Local Alignment Search Tool
 Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
A NEW GENERATION OF HOMOLOGY SEARCH TOOLS BASED ON PROBABILISTIC INFERENCE
 205 A NEW GENERATION OF HOMOLOGY SEARCH TOOLS BASED ON PROBABILISTIC INFERENCE SEAN R. EDDY 1 eddys@janelia.hhmi.org 1 Janelia Farm Research Campus, Howard Hughes Medical Institute, 19700 Helix Drive,
205 A NEW GENERATION OF HOMOLOGY SEARCH TOOLS BASED ON PROBABILISTIC INFERENCE SEAN R. EDDY 1 eddys@janelia.hhmi.org 1 Janelia Farm Research Campus, Howard Hughes Medical Institute, 19700 Helix Drive,
Lab 4: Multiple Sequence Alignment (MSA)
") Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
1. R. Durbin, S. Eddy, A. Krogh und G. Mitchison: Biological sequence analysis, Cambridge, 1998
 7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
Multiple sequence alignment. November 2, 2017
 Multiple sequence alignment November 2, 2017 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one sequence
Multiple sequence alignment November 2, 2017 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one sequence
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
PFstats User Guide. Aspartate/ornithine carbamoyltransferase Case Study. Neli Fonseca
 PFstats User Guide Aspartate/ornithine carbamoyltransferase Case Study 1 Contents Overview 3 Obtaining An Alignment 3 Methods 4 Alignment Filtering............................................ 4 Reference
PFstats User Guide Aspartate/ornithine carbamoyltransferase Case Study 1 Contents Overview 3 Obtaining An Alignment 3 Methods 4 Alignment Filtering............................................ 4 Reference
Documentation of HMMEditor 1.0
 Documentation of HMMEditor 1.0 HMMEditor 1.0 stands for profile Hidden Markov Model (phmm) Visual Editor. It is a tool to visualize and edit phmm in HMMer format. HMMer format is also used by Pfam protein
Documentation of HMMEditor 1.0 HMMEditor 1.0 stands for profile Hidden Markov Model (phmm) Visual Editor. It is a tool to visualize and edit phmm in HMMer format. HMMer format is also used by Pfam protein
Genome 559. Hidden Markov Models
 Genome 559 Hidden Markov Models A simple HMM Eddy, Nat. Biotech, 2004 Notes Probability of a given a state path and output sequence is just product of emission/transition probabilities If state path is
Genome 559 Hidden Markov Models A simple HMM Eddy, Nat. Biotech, 2004 Notes Probability of a given a state path and output sequence is just product of emission/transition probabilities If state path is
Gibbs Sampling. Stephen F. Altschul. National Center for Biotechnology Information National Library of Medicine National Institutes of Health
 Gibbs Sampling Stephen F. Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Optimization in High-Dimensional Space Smooth and simple landscapes
Gibbs Sampling Stephen F. Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Optimization in High-Dimensional Space Smooth and simple landscapes
A Layer-Based Approach to Multiple Sequences Alignment
 A Layer-Based Approach to Multiple Sequences Alignment Tianwei JIANG and Weichuan YU Laboratory for Bioinformatics and Computational Biology, Department of Electronic and Computer Engineering, The Hong
A Layer-Based Approach to Multiple Sequences Alignment Tianwei JIANG and Weichuan YU Laboratory for Bioinformatics and Computational Biology, Department of Electronic and Computer Engineering, The Hong
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Lecture 4: January 1, Biological Databases and Retrieval Systems
 Algorithms for Molecular Biology Fall Semester, 1998 Lecture 4: January 1, 1999 Lecturer: Irit Orr Scribe: Irit Gat and Tal Kohen 4.1 Biological Databases and Retrieval Systems In recent years, biological
Algorithms for Molecular Biology Fall Semester, 1998 Lecture 4: January 1, 1999 Lecturer: Irit Orr Scribe: Irit Gat and Tal Kohen 4.1 Biological Databases and Retrieval Systems In recent years, biological
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity
 A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity G. Pratyusha, Department of Computer Science & Engineering, V.R.Siddhartha Engineering College(Autonomous)
A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity G. Pratyusha, Department of Computer Science & Engineering, V.R.Siddhartha Engineering College(Autonomous)
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Basics of Multiple Sequence Alignment
 Basics of Multiple Sequence Alignment Tandy Warnow February 10, 2018 Basics of Multiple Sequence Alignment Tandy Warnow Basic issues What is a multiple sequence alignment? Evolutionary processes operating
Basics of Multiple Sequence Alignment Tandy Warnow February 10, 2018 Basics of Multiple Sequence Alignment Tandy Warnow Basic issues What is a multiple sequence alignment? Evolutionary processes operating
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships
 Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
Lecture 10: Local Alignments
 Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Two Phase Evolutionary Method for Multiple Sequence Alignments
 The First International Symposium on Optimization and Systems Biology (OSB 07) Beijing, China, August 8 10, 2007 Copyright 2007 ORSC & APORC pp. 309 323 Two Phase Evolutionary Method for Multiple Sequence
The First International Symposium on Optimization and Systems Biology (OSB 07) Beijing, China, August 8 10, 2007 Copyright 2007 ORSC & APORC pp. 309 323 Two Phase Evolutionary Method for Multiple Sequence
A multiple alignment tool in 3D
 Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
FastA & the chaining problem
 FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD
 Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:
 FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Dynamic Programming: Sequence alignment. CS 466 Saurabh Sinha
 Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Biology 644: Bioinformatics
 A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
Local weighting schemes for protein multiple sequence alignment
 Computers and Chemistry 26 (2002) 459 477 www.elsevier.com/locate/compchem Local weighting schemes for protein multiple sequence alignment Jaap Heringa 1 * Di ision of Mathematical Biology, MRC National
Computers and Chemistry 26 (2002) 459 477 www.elsevier.com/locate/compchem Local weighting schemes for protein multiple sequence alignment Jaap Heringa 1 * Di ision of Mathematical Biology, MRC National
A CAM(Content Addressable Memory)-based architecture for molecular sequence matching
-based architecture for molecular sequence matching") A CAM(Content Addressable Memory)-based architecture for molecular sequence matching P.K. Lala 1 and J.P. Parkerson 2 1 Department Electrical Engineering, Texas A&M University, Texarkana, Texas, USA 2
A CAM(Content Addressable Memory)-based architecture for molecular sequence matching P.K. Lala 1 and J.P. Parkerson 2 1 Department Electrical Engineering, Texas A&M University, Texarkana, Texas, USA 2
Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences
 Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences Yue Lu and Sing-Hoi Sze RECOMB 2007 Presented by: Wanxing Xu March 6, 2008 Content Biology Motivation Computation Problem
Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences Yue Lu and Sing-Hoi Sze RECOMB 2007 Presented by: Wanxing Xu March 6, 2008 Content Biology Motivation Computation Problem
Multiple Sequence Alignment. With thanks to Eric Stone and Steffen Heber, North Carolina State University
 Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
15-780: Graduate Artificial Intelligence. Computational biology: Sequence alignment and profile HMMs
 5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
Multiple alignment. Multiple alignment. Computational complexity. Multiple sequence alignment. Consensus. 2 1 sec sec sec
 Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu VTISCTGSSSNIGAG-NHVKWYQQLPG VTISCTGTSSNIGS--ITVNWYQQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG PEVTCVVVDVSHEDPQVKFNWYVDG--
Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu VTISCTGSSSNIGAG-NHVKWYQQLPG VTISCTGTSSNIGS--ITVNWYQQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG PEVTCVVVDVSHEDPQVKFNWYVDG--
USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT
 IADIS International Conference Applied Computing 2006 USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT Divya R. Singh Software Engineer Microsoft Corporation, Redmond, WA 98052, USA Abdullah
IADIS International Conference Applied Computing 2006 USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT Divya R. Singh Software Engineer Microsoft Corporation, Redmond, WA 98052, USA Abdullah
Gene regulation. DNA is merely the blueprint Shared spatially (among all tissues) and temporally But cells manage to differentiate
 and temporally But cells manage to differentiate") Gene regulation DNA is merely the blueprint Shared spatially (among all tissues) and temporally But cells manage to differentiate Especially but not only during developmental stage And cells respond to
Gene regulation DNA is merely the blueprint Shared spatially (among all tissues) and temporally But cells manage to differentiate Especially but not only during developmental stage And cells respond to
PaMSA: A Parallel Algorithm for the Global Alignment of Multiple Protein Sequences
 PaMSA: A Parallel Algorithm for the Global Alignment of Multiple Protein Sequences Irma R. Andalon-Garcia and Arturo Chavoya Department of Information Systems Guadalajara University CUCEA Guadalajara,
PaMSA: A Parallel Algorithm for the Global Alignment of Multiple Protein Sequences Irma R. Andalon-Garcia and Arturo Chavoya Department of Information Systems Guadalajara University CUCEA Guadalajara,