Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties
|
|
|
- Hope Barker
- 6 years ago
- Views:
Transcription
1
2 Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties
3 From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence alignment allows only insertions and deletions (no mismatches). In the LCS Problem, we scored 1 for matches and 0 for indels Consider penalizing indels and mismatches with negative scores Simplest scoring schema: +1 : match premium -µ : mismatch penalty -σ : indel penalty
4 When mismatches are penalized by µ, indels are penalized by σ, and matches are rewarded with +1, the resulting score is: #matches µ(#mismatches) σ(#indels)
5 Find the best alignment between two strings under a given scoring schema Input : Strings v and w and a scoring schema Output : Alignment of maximum score = -б = 1 if match = -µ if mismatch s i-1,j-1 +1 if v i = w j µ : mismatch penalty s i,j = max s i-1,j-1 -µ if v i w j s i-1,j -σ s i,j-1 -σ σ : indel penalty
6 To generalize scoring, consider a (4+1) x(4+1) scoring matrixδ. In the case of an amino acid sequence alignment, the scoring matrix would be a (20+1)x(20+1) size. The addition of 1 is to include the score for comparison of a gap character -. This will simplify the algorithm as follows: s + δ (v, w ) i-1,j-1 i j s = max s + δ (v, -) i,j i-1,j i s + δ (-, w ) i,j-1 j
7 Measuring the extent of similarity between two sequences Based on percent sequence identity Based on conservation
8 The extent to which two nucleotide or amino acid sequences are invariant mismatch 70% identical indel
9 Scoring matrices are created based on biological evidence. Alignments can be thought of as two sequences that differ due to mutations. Some of these mutations have little effect on the protein s function, therefore some penalties, δ(v, w ), will be less harsh than i j others.
10 A R N K Notice that although A R and K are different R amino acids, they N have a positive score. K Why? They are both positively charged amino acids will not greatly change function of protein.
11 Amino acid changes that tend to preserve the physico-chemical properties of the original residue Polar to polar aspartate glutamate Nonpolar to nonpolar alanine valine Similarly behaving residues leucine to isoleucine
12 Amino acid substitution matrices PAM BLOSUM DNA substitution matrices DNA is less conserved than protein sequences Less effective to compare coding regions at nucleotide level
13 Point Accepted Mutation (Dayhoff et al.) 1 PAM = PAM = 1% average change of all amino 1 acid positions After 100 PAMs of evolution, not every residue will have changed some residues may have mutated several times some residues may have returned to their original state some residues may not changed at all
14 X PAM x = PAM 1 x PAM 250 = PAM PAM 250 is a common PAM scoring matrix: Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys... A R N D C Q E G H I L K... Ala A Arg R Asn N Asp D Cys C Gln Q Trp W Tyr Y Val V
15 Blocks Substitution Matrix Scores derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins Matrix name indicates evolutionary distance BLOSUM62 was created using sequences sharing no more than 62% identity
 X=Any")
16 B=N/D=Asx (Asn/Asp) Z=Q/E=Glx (Gln,Glu) X=Any AA J=I/L
17 The Global Alignment Problem tries to find the longest path between vertices (0,0) and (n,m) in the edit graph. The Local Alignment Problem tries to find the longest path among paths between arbitrary vertices (i,j) and (i, j ) in the edit graph.
18 The Global Alignment Problem tries to find the longest path between vertices (0,0) and (n,m) in the edit graph. The Local Alignment Problem tries to find the longest path among paths between arbitrary vertices (i,j) and (i, j ) in the edit graph. In the edit graph with negatively-scored edges, Local Alignment may score higher than Global Alignment
19 (cont d) Global Alignment --T -CC-C-AGT -TATGT-CAGGGGACACG A-GCATGCAGA-GAC AATTGCCGCC-GTCGT-T-TTCAG----CA-GTTATG T-CAGAT--C Local Alignment better alignment to find conserved segment tcccagttatgtcaggggacacgagcatgcagagac aattgccgccgtcgttttcagcagttatgtcagatc
20 Compute a mini Local alignment Global Alignment to get Local Global alignment
21 Two genes in different species may be similar over short conserved regions and dissimilar over remaining regions. Example: Homeobox genes have a short region called the homeodomain that is highly conserved between species. A global alignment would not find the homeodomain because it would try to align the ENTIRE sequence
22 Goal: Find the best local alignment between two strings Input : Strings v, w and scoring matrix δ Output : Alignment of substrings of v and w whose alignment score is maximum among all possible alignment of all possible substrings
23 Long run time O(n 4 ): - In the grid of size n x n there are ~n 2 vertices (i,j) that may serve as a source. - For each such vertex computing alignments from (i,j) to (i,j ) takes O(n 2 ) time.
24 Compute a mini Local alignment Global Alignment to get Local Global alignment
25
26
27
28
29
30 Long run time O(n 4 ): - In the grid of size n x n there are ~n 2 vertices (i,j) that may serve as a source. - For each such vertex computing alignments from (i,j) to (i,j ) takes O(n 2 ) time. This can be remedied by giving free rides
 The dashed edges represent the free")
31 Yeah, a free ride! Vertex (0,0) The dashed edges represent the free rides from (0,0) to every other node.
32 The largest value of s i,j over the whole edit graph is the score of the best local alignment. The recurrence: 0 s i,j = max s i-1,j-1 + δ (v i, w j ) s i-1,j + δ (v i, -) Notice there is only this change from the original recurrence of a Global Alignment s i,j-1 + δ (-, w j )
33 The largest value of s i,j over the whole edit graph is the score of the best local alignment. The recurrence: Power of ZERO: there is 0 s = max s + δ (v, w ) i,j i-1,j-1 i j s + δ (v, -) i-1,j i s + δ (-, w ) i,j-1 j only this change from the original recurrence of a Global Alignment - since there is only one free ride edge entering into every vertex
34 A fixed penalty σ is given to every indel: -σ for 1 indel, -2σ for 2 consecutive indels -3σ for 3 consecutive indels, etc. Can be too severe penalty for a series of 100 consecutive indels
35 In nature, a series of k indels often come as a single event rather than a series of k single nucleotide events: Normal scoring would This is more likely. give the same score for both alignments This is less likely.
36 Gaps- contiguous sequence of spaces in one of the rows Score for a gap of length x is: -(ρ +σx) where ρ >0 is the penalty for introducing a gap: gap opening penalty ρ will be large relative to σ: gap extension penalty because you do not want to add too much of a penalty for extending the gap.
37 Gap penalties: -ρ-σ when there is 1 indel -ρ-2σ when there are 2 indels -ρ-3σ when there are 3 indels, etc. -ρ- x σ (-gap opening - x gap extensions) Somehow reduced penalties (as compared to naïve scoring) are given to runs of horizontal and vertical edges
38 To reflect affine gap penalties we have to add long horizontal and vertical edges to the edit graph. Each such edge of length x should have weight -ρ - x *σ
39 Adding Affine Penalty Edges to the Edit Graph There are many such edges! Adding them to the graph increases the running time of the alignment algorithm by a factor of n (where n is the number of vertices) So the complexity increases from O(n 2 ) to O(n 3 )
40 ρ δ δ σ δ ρ δ δ σ
41 Affine Gap Penalties and 3 Layer Manhattan Grid The three recurrences for the scoring algorithm creates a 3-layered graph. The top level creates/extends gaps in the sequence w. The bottom level creates/extends gaps in sequence v. The middle level extends matches and mismatches.
42 Levels: The main level is for diagonal edges The lower level is for horizontal edges The upper level is for vertical edges A jumping penalty is assigned to moving from the main level to either the upper level or the lower level (-ρ σ) There is a gap extension penalty for each continuation on a level other than the main level (-σ)
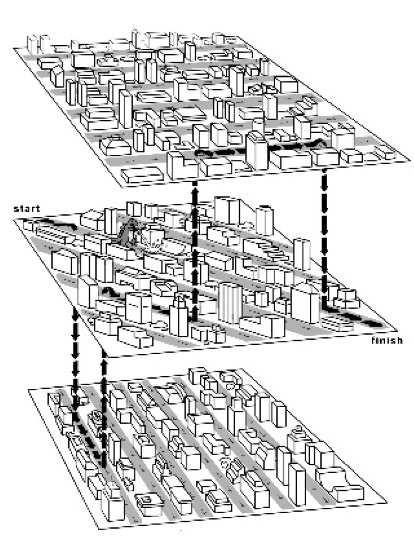
43
44 s i,j = s i-1,j -σ max s i-1,j (ρ+σ) Continue Gap in w (deletion) Start Gap in w (deletion): from middle s i,j = s i,j-1 -σ max s i,j-1 (ρ+σ) Continue Gap in v (insertion) Start Gap in v (insertion):from middle s i,j = s i-1,j-1 + δ (v i, w j ) max s i,j Match or Mismatch End deletion: from top s i,j End insertion: from bottom
Dynamic Programming: Sequence alignment. CS 466 Saurabh Sinha
 Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS 4425: Introductory Computational Bioinformatics Fall Suprakash Datta
 EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Lecture 10: Local Alignments
 Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Algorithmic Approaches for Biological Data, Lecture #20
 Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
6.1 The Power of DNA Sequence Comparison
 6 Dynamic Programming Algorithms We introduced dynamic programming in chapter 2 with the Rocks problem. While the Rocks problem does not appear to be related to bioinformatics, the algorithm that we described
6 Dynamic Programming Algorithms We introduced dynamic programming in chapter 2 with the Rocks problem. While the Rocks problem does not appear to be related to bioinformatics, the algorithm that we described
Greedy Algorithms Huffman Coding
 Greedy Algorithms Huffman Coding Huffman Coding Problem Example: Release 29.1 of 15-Feb-2005 of TrEMBL Protein Database contains 1,614,107 sequence entries, comprising 505,947,503 amino acids. There are
Greedy Algorithms Huffman Coding Huffman Coding Problem Example: Release 29.1 of 15-Feb-2005 of TrEMBL Protein Database contains 1,614,107 sequence entries, comprising 505,947,503 amino acids. There are
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD
 Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Building and Animating Amino Acids and DNA Nucleotides in ShockWave Using 3ds max
 1 Building and Animating Amino Acids and DNA Nucleotides in ShockWave Using 3ds max MIT Center for Educational Computing Initiatives THIS PDF DOCUMENT HAS BOOKMARKS FOR NAVIGATION CLICK ON THE TAB TO THE
1 Building and Animating Amino Acids and DNA Nucleotides in ShockWave Using 3ds max MIT Center for Educational Computing Initiatives THIS PDF DOCUMENT HAS BOOKMARKS FOR NAVIGATION CLICK ON THE TAB TO THE
Alignment of Pairs of Sequences
 Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Giri Narasimhan & Kip Irvine
 COP 4516: Competitive Programming and Problem Solving! Giri Narasimhan & Kip Irvine Phone: x3748 & x1528 {giri,irvinek}@cs.fiu.edu Problems to think about!! What is the least number of comparisons you
COP 4516: Competitive Programming and Problem Solving! Giri Narasimhan & Kip Irvine Phone: x3748 & x1528 {giri,irvinek}@cs.fiu.edu Problems to think about!! What is the least number of comparisons you
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Lecture 3: February Local Alignment: The Smith-Waterman Algorithm
 CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
Assignment 4. the three-dimensional positions of every single atom in the le,
 Assignment 4 1 Overview and Background Many of the assignments in this course will introduce you to topics in computational biology. You do not need to know anything about biology to do these assignments
Assignment 4 1 Overview and Background Many of the assignments in this course will introduce you to topics in computational biology. You do not need to know anything about biology to do these assignments
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
DNA Alignment With Affine Gap Penalties
 DNA Alignment With Affine Gap Penalties Laurel Schuster Why Use Affine Gap Penalties? When aligning two DNA sequences, one goal may be to infer the mutations that made them different. Though it s impossible
DNA Alignment With Affine Gap Penalties Laurel Schuster Why Use Affine Gap Penalties? When aligning two DNA sequences, one goal may be to infer the mutations that made them different. Though it s impossible
RB-Tree Augmentation. OS-Rank. OS-Select. Augment x with Size(x), where. Size(x) = size of subtree rooted at x Size(NIL) = 0
, where. Size(x) = size of subtree rooted at x Size(NIL) = 0") RB-Tree Augmentation Augment x with Size(x), where Size(x) = size of subtree rooted at x Size(NIL) = 0 COT 5407 10/6/05 1 OS-Rank OS-RANK(x,y) // Different from text (recursive version) // Find the rank
RB-Tree Augmentation Augment x with Size(x), where Size(x) = size of subtree rooted at x Size(NIL) = 0 COT 5407 10/6/05 1 OS-Rank OS-RANK(x,y) // Different from text (recursive version) // Find the rank
Multiple Sequence Alignment. Mark Whitsitt - NCSA
 Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
PROTEIN MULTIPLE ALIGNMENT MOTIVATION: BACKGROUND: Marina Sirota
 Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Dynamic Programming Part I: Examples. Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, / 77
 Dynamic Programming -class4- July 25th, / 77") Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Today s Lecture. Edit graph & alignment algorithms. Local vs global Computational complexity of pairwise alignment Multiple sequence alignment
 Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Supporting information
 Supporting information 1S. DECOMP output for the peptide amino-acid decomposition test with monoisotopic mass = 1000 +/- 0.2 # imsdecomp 1.3 # Copyright 2007,2008 Informatics for Mass Spectrometry group
Supporting information 1S. DECOMP output for the peptide amino-acid decomposition test with monoisotopic mass = 1000 +/- 0.2 # imsdecomp 1.3 # Copyright 2007,2008 Informatics for Mass Spectrometry group
Shortest Path Algorithm
 Shortest Path Algorithm C Works just fine on this graph. C Length of shortest path = Copyright 2005 DIMACS BioMath Connect Institute Robert Hochberg Dynamic Programming SP #1 Same Questions, Different
Shortest Path Algorithm C Works just fine on this graph. C Length of shortest path = Copyright 2005 DIMACS BioMath Connect Institute Robert Hochberg Dynamic Programming SP #1 Same Questions, Different
Machine Learning Methods. Majid Masso, PhD Bioinformatics and Computational Biology George Mason University
 Machine Learning Methods Majid Masso, PhD Bioinformatics and Computational Biology George Mason University Introductory Example Attributes X and Y measured for each person (example or instance) in a training
Machine Learning Methods Majid Masso, PhD Bioinformatics and Computational Biology George Mason University Introductory Example Attributes X and Y measured for each person (example or instance) in a training
Supplementary Information
 Supplementary Information Supplementary Figure S1 The scheme of MtbHadAB/MtbHadBC dehydration reaction. The reaction is reversible. However, in the context of FAS-II elongation cycle, this reaction tends
Supplementary Information Supplementary Figure S1 The scheme of MtbHadAB/MtbHadBC dehydration reaction. The reaction is reversible. However, in the context of FAS-II elongation cycle, this reaction tends
- G T G T A C A C
 Name Student ID.. Sequence alignment 1. Globally align sequence V (GTGTACAC) and sequence W (GTACC) by hand using dynamic programming algorithm. The alignment will be performed based on match premium of
Name Student ID.. Sequence alignment 1. Globally align sequence V (GTGTACAC) and sequence W (GTACC) by hand using dynamic programming algorithm. The alignment will be performed based on match premium of
Dynamic Programming and Applications
 Dynamic Programming and Applications Michael Schatz Bioinformatics Lecture 2 Quantitative Biology 2013 Exact Matching Review Where is GATTACA in the human genome? E=183,105 Brute Force (3 GB) Suffix Array
Dynamic Programming and Applications Michael Schatz Bioinformatics Lecture 2 Quantitative Biology 2013 Exact Matching Review Where is GATTACA in the human genome? E=183,105 Brute Force (3 GB) Suffix Array
Sequence alignment algorithms
 Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Similarity Searches on Sequence Databases
 Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Lab 4: Multiple Sequence Alignment (MSA)
") Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Notes on Dynamic-Programming Sequence Alignment
 Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
SAY IT WITH DNA: Making New Messages
 Y WH : Making New Messages ince you will be expected to decipher a message in the unit exam, it would be wise to get as much practice as possible. f you can have fun in the process, so much the better!
Y WH : Making New Messages ince you will be expected to decipher a message in the unit exam, it would be wise to get as much practice as possible. f you can have fun in the process, so much the better!
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Pairwise Sequence Alignment: Dynamic Programming Algorithms. COMP Spring 2015 Luay Nakhleh, Rice University
 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
Lecture 9: Core String Edits and Alignments
 Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Amino Acid Graph Representation for Efficient Safe Transfer of Multiple DNA Sequence as Pre Order Trees
 International Journal of Bioinformatics and Biomedical Engineering Vol. 1, No. 3, 2015, pp. 292-299 http://www.aiscience.org/journal/ijbbe Amino Acid Graph Representation for Efficient Safe Transfer of
International Journal of Bioinformatics and Biomedical Engineering Vol. 1, No. 3, 2015, pp. 292-299 http://www.aiscience.org/journal/ijbbe Amino Acid Graph Representation for Efficient Safe Transfer of
Sequence Alignment. Ulf Leser
 Sequence Alignment Ulf Leser his Lecture Approximate String Matching Edit distance and alignment Computing global alignments Local alignment Ulf Leser: Bioinformatics, Summer Semester 2016 2 ene Function
Sequence Alignment Ulf Leser his Lecture Approximate String Matching Edit distance and alignment Computing global alignments Local alignment Ulf Leser: Bioinformatics, Summer Semester 2016 2 ene Function
DynamicProgramming. September 17, 2018
 DynamicProgramming September 17, 2018 1 Lecture 11: Dynamic Programming CBIO (CSCI) 4835/6835: Introduction to Computational Biology 1.1 Overview and Objectives We ve so far discussed sequence alignment
DynamicProgramming September 17, 2018 1 Lecture 11: Dynamic Programming CBIO (CSCI) 4835/6835: Introduction to Computational Biology 1.1 Overview and Objectives We ve so far discussed sequence alignment
Research Article Aligning Sequences by Minimum Description Length
 Hindawi Publishing Corporation EURASIP Journal on Bioinformatics and Systems Biology Volume 2007, Article ID 72936, 14 pages doi:10.1155/2007/72936 Research Article Aligning Sequences by Minimum Description
Hindawi Publishing Corporation EURASIP Journal on Bioinformatics and Systems Biology Volume 2007, Article ID 72936, 14 pages doi:10.1155/2007/72936 Research Article Aligning Sequences by Minimum Description
Lecture 12: Divide and Conquer Algorithms
 Lecture 12: Divide and Conquer Algorithms Study Chapter 7.1 7.4 1 Divide and Conquer Algorithms Divide problem into sub-problems Conquer by solving sub-problems recursively. If the sub-problems are small
Lecture 12: Divide and Conquer Algorithms Study Chapter 7.1 7.4 1 Divide and Conquer Algorithms Divide problem into sub-problems Conquer by solving sub-problems recursively. If the sub-problems are small
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
Important Example: Gene Sequence Matching. Corrigiendum. Central Dogma of Modern Biology. Genetics. How Nucleotides code for Amino Acids
 Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Dynamic Programming & Smith-Waterman algorithm
 m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
Parameterized Complexity and Biopolymer Sequence Comparison
 Parameterized Complexity and Biopolymer Sequence Comparison Liming Cai, Xiuzhen Huang, Chunmei Liu, Frances Rosamond and Yinglei Song Abstract The article surveys parameterized algorithms and complexities
Parameterized Complexity and Biopolymer Sequence Comparison Liming Cai, Xiuzhen Huang, Chunmei Liu, Frances Rosamond and Yinglei Song Abstract The article surveys parameterized algorithms and complexities
3/5/2018 Lecture15. Comparing Sequences. By Miguel Andrade at English Wikipedia.
 3/5/2018 Lecture15 Comparing Sequences By Miguel Andrade at English Wikipedia 1 http://localhost:8888/notebooks/comp555s18/lecture15.ipynb# 1/1 Sequence Similarity A common problem in Biology Insulin Protein
3/5/2018 Lecture15 Comparing Sequences By Miguel Andrade at English Wikipedia 1 http://localhost:8888/notebooks/comp555s18/lecture15.ipynb# 1/1 Sequence Similarity A common problem in Biology Insulin Protein
Scoring and heuristic methods for sequence alignment CG 17
 Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Ramachandran Plot. 4ytn. PRO 51 (D) ~l. l TRP 539 (E) Phi (degrees) Plot statistics
 ~l. l TRP 539 (E) Phi (degrees) Plot statistics") B Ramachandran Plot ~b b 135 b ~b PRO 51 (D) ~l l TRP 539 (E) Psi (degrees) 5-5 a SER (B) A ~a L LYS (F) ALA 35 (E) - -135 ~b b HIS 59 (G) ALA 173 (E) ASP ALA 13173 (F)(A) ASP LYS 13315 LYS (B)(E) 315
B Ramachandran Plot ~b b 135 b ~b PRO 51 (D) ~l l TRP 539 (E) Psi (degrees) 5-5 a SER (B) A ~a L LYS (F) ALA 35 (E) - -135 ~b b HIS 59 (G) ALA 173 (E) ASP ALA 13173 (F)(A) ASP LYS 13315 LYS (B)(E) 315
Divya R. Singh. Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques. February A Thesis Presented by
 Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques A Thesis Presented by Divya R. Singh to The Faculty of the Graduate College of the University of Vermont In Partial Fulfillment of
Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques A Thesis Presented by Divya R. Singh to The Faculty of the Graduate College of the University of Vermont In Partial Fulfillment of
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Sequence Alignment: Mo1va1on and Algorithms. Lecture 2: August 23, 2012
 Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Divide & Conquer Algorithms
 Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Sequence Alignment Heuristics
 Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
A Bit-Parallel, General Integer-Scoring Sequence Alignment Algorithm
 A Bit-Parallel, General Integer-Scoring Sequence Alignment Algorithm GARY BENSON, YOZEN HERNANDEZ, & JOSHUA LOVING B I O I N F O R M A T I C S P R O G R A M B O S T O N U N I V E R S I T Y J L O V I N
A Bit-Parallel, General Integer-Scoring Sequence Alignment Algorithm GARY BENSON, YOZEN HERNANDEZ, & JOSHUA LOVING B I O I N F O R M A T I C S P R O G R A M B O S T O N U N I V E R S I T Y J L O V I N
Homework Python-1. Sup Biotech 3 Python. Pierre Parutto
 Homework Python-1 Sup Biotech 3 Python Pierre Parutto October 9, 2016 Preamble Document Property Authors Pierre Parutto Version 1.0 Number of pages 9 Contact Contact the assistant team at: supbiotech-bioinfo-bt3@googlegroups.com
Homework Python-1 Sup Biotech 3 Python Pierre Parutto October 9, 2016 Preamble Document Property Authors Pierre Parutto Version 1.0 Number of pages 9 Contact Contact the assistant team at: supbiotech-bioinfo-bt3@googlegroups.com
Sequence Alignment. COMPSCI 260 Spring 2016
 Sequence Alignment COMPSCI 260 Spring 2016 Why do we want to compare DNA or protein sequences? Find genes similar to known genes IdenGfy important (funcgonal) sequences by finding conserved regions As
Sequence Alignment COMPSCI 260 Spring 2016 Why do we want to compare DNA or protein sequences? Find genes similar to known genes IdenGfy important (funcgonal) sequences by finding conserved regions As
Homework Python-1. Sup Biotech 3 Python. Pierre Parutto
 Homework Python-1 Sup Biotech 3 Python Pierre Parutto November 7, 2016 Preamble Document Property Authors Pierre Parutto Version 1.0 Number of pages 14 Contact Contact the assistant team at: supbiotech-bioinfo-bt3@googlegroups.com
Homework Python-1 Sup Biotech 3 Python Pierre Parutto November 7, 2016 Preamble Document Property Authors Pierre Parutto Version 1.0 Number of pages 14 Contact Contact the assistant team at: supbiotech-bioinfo-bt3@googlegroups.com
Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--
 Sequence Alignment Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC-- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Distance from sequences
Sequence Alignment Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC-- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Distance from sequences
BLAST - Basic Local Alignment Search Tool
 Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
1. Open the SPDBV_4.04_OSX folder on the desktop and double click DeepView to open.
 Molecular of inhibitor-bound Lysozyme This lab will not require a lab report. Rather each student will follow this tutorial, answer the italicized questions (worth 2 points each) directly on this protocol/worksheet,
Molecular of inhibitor-bound Lysozyme This lab will not require a lab report. Rather each student will follow this tutorial, answer the italicized questions (worth 2 points each) directly on this protocol/worksheet,
Weighted Finite-State Transducers in Computational Biology
 Weighted Finite-State Transducers in Computational Biology Mehryar Mohri Courant Institute of Mathematical Sciences mohri@cims.nyu.edu Joint work with Corinna Cortes (Google Research). 1 This Tutorial
Weighted Finite-State Transducers in Computational Biology Mehryar Mohri Courant Institute of Mathematical Sciences mohri@cims.nyu.edu Joint work with Corinna Cortes (Google Research). 1 This Tutorial
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University
 1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
Profiles and Multiple Alignments. COMP 571 Luay Nakhleh, Rice University
 Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Finding sequence similarities with genes of known function is a common approach to infer a newly sequenced gene s function
 Outline DNA Sequence Comparison: First Success Stories Change Problem Manhattan Tourist Problem Longest Paths in Graphs Sequence Alignment Edit Distance Longest Common Subsequence Problem Dot Matrices
Outline DNA Sequence Comparison: First Success Stories Change Problem Manhattan Tourist Problem Longest Paths in Graphs Sequence Alignment Edit Distance Longest Common Subsequence Problem Dot Matrices
Sequence Analysis '17: lecture 4. Molecular evolution Global, semi-global and local Affine gap penalty
 Sequence Analysis '17: lecture 4 Molecular evolution Global, semi-global and local Affine gap penalty How sequences evolve point mutations (single base changes) deletion (loss of residues within the sequence)
Sequence Analysis '17: lecture 4 Molecular evolution Global, semi-global and local Affine gap penalty How sequences evolve point mutations (single base changes) deletion (loss of residues within the sequence)
Parameterized Complexity and Biopolymer Sequence Comparison
 The Computer Journal Advance Access published June 27, 2007 # The Author 2007. Published by Oxford University Press on behalf of The British Computer Society. All rights reserved. For Permissions, please
The Computer Journal Advance Access published June 27, 2007 # The Author 2007. Published by Oxford University Press on behalf of The British Computer Society. All rights reserved. For Permissions, please
Dynamic Programming Comp 122, Fall 2004
 Dynamic Programming Comp 122, Fall 2004 Review: the previous lecture Principles of dynamic programming: optimization problems, optimal substructure property, overlapping subproblems, trade space for time,
Dynamic Programming Comp 122, Fall 2004 Review: the previous lecture Principles of dynamic programming: optimization problems, optimal substructure property, overlapping subproblems, trade space for time,
A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity
 A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity G. Pratyusha, Department of Computer Science & Engineering, V.R.Siddhartha Engineering College(Autonomous)
A Study On Pair-Wise Local Alignment Of Protein Sequence For Identifying The Structural Similarity G. Pratyusha, Department of Computer Science & Engineering, V.R.Siddhartha Engineering College(Autonomous)
Pairwise Sequence alignment Basic Algorithms
 Pairwise Sequence alignment Basic Algorithms Agenda - Previous Lesson: Minhala - + Biological Story on Biomolecular Sequences - + General Overview of Problems in Computational Biology - Reminder: Dynamic
Pairwise Sequence alignment Basic Algorithms Agenda - Previous Lesson: Minhala - + Biological Story on Biomolecular Sequences - + General Overview of Problems in Computational Biology - Reminder: Dynamic
Divide & Conquer Algorithms
 Divide & Conquer Algorithms Outline 1. MergeSort 2. Finding the middle vertex 3. Linear space sequence alignment 4. Block alignment 5. Four-Russians speedup 6. LCS in sub-quadratic time Section 1: MergeSort
Divide & Conquer Algorithms Outline 1. MergeSort 2. Finding the middle vertex 3. Linear space sequence alignment 4. Block alignment 5. Four-Russians speedup 6. LCS in sub-quadratic time Section 1: MergeSort
Python for Bioinformatics
 Python for Bioinformatics A look into the BioPython world... Christian Skjødt csh@cbs.dtu.dk Today s program 09-10: 10-12: 12-13: 13-13.30: 13.30-16: 16:00-17: Lecture: Introduction to BioPython Exercise:
Python for Bioinformatics A look into the BioPython world... Christian Skjødt csh@cbs.dtu.dk Today s program 09-10: 10-12: 12-13: 13-13.30: 13.30-16: 16:00-17: Lecture: Introduction to BioPython Exercise:
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding. B. Majoros M. Pertea S.L. Salzberg
 Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
Lecture 5: Markov models
 Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Multiple Sequence Alignment (MSA)
") I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment
 Multiple Sequence Alignment") GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic