Preliminary Syllabus. Genomics. Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification
|
|
|
- Joel Carson
- 6 years ago
- Views:
Transcription
1 Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK Comparative Protein-Protein Interactions Pathway Resources and Analysis Structural / Protein Structure Prediction Protein Modeling EXAM Gribskov@purdue.edu Lilly G-233 Gribskov 2.1
2 Genome Assembly Populus trichocarpa Science (2006) 313: (September 15) 485 Mb (cytogenetic estimate = 550 Mb) 7.5X coverage 2447 scaffolds, 410Mb in scaffold assembly (84%) 95% of genome 45,500 "genes" 19 Linkage groups Evidence for two whole genome duplications Gribskov 2.2
3 Genome Assembly Populus Clone and sequence statistics ti ti Insert Size Kb Vector Number Reads x10-6 Number Reads Number Bases Used Qual > 20 x10-6 Gb Number Bases After Trimming % Bases Used % of Total Gb plasmid plasmid fosmid 0, Total Gribskov 2.3


4 Genome Assembly Populus Small contigs and singletons tend to be contaminants Gribskov 2.4
 (black) shows clone coverage, Each")
 shows the coverage provided by clones not")
 shows anchored contigs Next")

5 Genome Assembly Populus Inner tracks (inside to outside ) (black) shows clone coverage, Each circle shows 5X depth. (red) shows the coverage provided by clones not assigned to contigs (singletons). (alternating color) shows anchored contigs Next (alternating color) track shows position of individual anchored clones in each contig Outer tracks clones lacking contig assignment. singletons. 1Mb Gribskov 2.5

6 Genome Assembly Populus How common are chimeras? Chimeric reads in Chloroplast l genome one end chloroplast one end nuclear Average 410 reads/position ~ 5-6% Gribskov 2.6

7 Genome Assembly Populus Transposable Elements Gribskov 2.7
8 Genome Assembly Additional Assembly Protocols Comparative assembly - Align to existing very similar to genome Several times faster 3-4X Problems Insertions/deletions Rearrangements Align by physical map Gribskov 2.8
9 EST Assembly Assembling RNA Result often called unigenes Much less consistent than DNA Similar to DNA except Contigs do not join into one sequence Special Artifacts Post-transcriptional modification Alternative splicing Trans-splicing SNPs/haplotypes Gribskov 2.9
10 Genome Assembly Assembly Validation how good is it? mate-pair information number of mate pairs whose distance violates length assumptions number of mate-pairs whose orientation is impossible see Phillipy et al., 2008 number of unused reads (singletons) align singletons to contigs to check correlated polymorphisms overlapping reads should not have differences at the same position unless mis-assembled allelic duplicated Experimental physical map FISH Gribskov 2.10




11 Genome Assembly Populus Mapping of scaffolds to chromosomes using microsatellites Gribskov 2.11


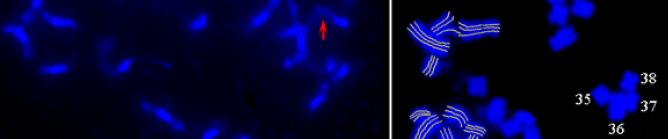

12 Genome Assembly Populus Mapping BACs to chromosomes using FISH Gribskov 2.12

13 Genome Assembly Mate-Pair violations Compressed tandem repeats make mate-pairs appear "stretched" incorrect arrangement of contigs leads to mis-oriented and inconsistent matepairs Gribskov 2.13

14 Genome Assembly Mate-Pair Violations B. anthacis example 4 unassembled regions partially match assembly partial matches all end at same location Gribskov 2.14


15 Genome Assembly Mate-Pair Drosophila virilis repeat compression insert in assembly? Gribskov 2.15
16 Genome Assembly Mate-pair Violations 16 Phrap bacterial genome assemblies Gribskov 2.16
17 Genome Assembly Finding Overlaps Most time consuming aspect of assembly Requires n 2 /2 comparisons = O(n 2 ) All methods rely on looking for exact matches over some length Two concerns How likely are incorrect matches How do to it very quickly Gribskov 2.17
18 Sequence Database Searching Essentially same problem as finding overlaps in assembly Main approach Rapid scan of database for candidate matches Slow evaluation of similarity il it by dynamic programming alignment Statistical analysis BLAST theory based FASTA fit to observed data Gribskov 2.18
19 Sequence database searching Gribskov 2.19
20 FASTA Originally developed in the mid-1980s as FASTN and FASTP for nucleic acid and protein, respectively Fast approximation of dynamic programming alignment Relies on related sequences having "diagonals" " with high h similarity il it Step 1. Find best regions on diagonals Step 2. Rescan 10 best regions with PAM scoring table Step 3. Join initial regions Step 4. Calculate dynamic programming optimal alignment Step 5. Calculate significance of Scores Gribskov 2.20
21 Sequence database searching - FASTA Step 1. Find best regions on diagonals Step 2. Rescan 10 best regions with scoring table Step 3. Join initial regions Step 4. Calculate dynamic programming optimal alignment Gribskov 2.21
22 Sequence database searching - FASTA Step 1 - Find Initial Regions (Fast part of search) Find best regions of diagonals using lookup table Lookup table: lists all the words of length ktup and where they occur Gribskov 2.22 MYSEQVENCEN HISSEQENCEQ CE 9 CE 9 EN 7,10 EN 7 EQ 4 EQ 5,10 MY 1 HI 1 NC 8 IS 2 QV 5 NC 8 SE 3 QE 6 UE 6 SE 4 YS 2 SS 3
23 Sequence database searching - FASTA Step 1 - Find Initial Regions For each matching word (ktup) calculate on which diagonal the match lies - AKA histograming diagonal = offset database - offset query CE 9 CE 9 0 EN 7,10 EN 7 0, +3 EQ 4 EQ 5,10-1, -6 MY 1 HI 1 0 Does it already have a region? If no, start a region (score=pair score) If yes, try to combine them score > distance to existing region (score = pair scores - distance) Gribskov 2.23
24 Sequence database searching Gribskov 2.24
25 Statistics Sequence matching is not normal, it is extreme! Scores follow and extreme value or Gumbel distribution Z score can't be directly converted to probability Whenever you are looking at a distribution of maxima longest run of heads in coin toss maximum scores for each sequence in database Sequence matches are a lot like coin tosses! PTVQGLRLFE :: : : PTAAGQELLS Gribskov 2.25
26 Extreme Value Distributions Are appropriate whenever you are looking at a DISTRIBUTION OF MAXIMA longest run of heads in coin toss maximum scores for each sequence in database Z score can't be directly converted to probability because it not a Normal or Gaussian distribution e.g. Z=3 has a normal P-value = but an extreme value distribution P-value ~ 0.12!!! about 100-fold error (error gets worse for smaller P-values)!!!!! Gribskov 2.26
27 Sequence Database Searching Score Distribution Cumulativ ve Probability Extreme Value Distribution Cumulative Probability 0.2 Probability 0.05 Gribskov Run Length 0
28 BLAST Based on Maximal Segment Pairs (MSP) Highest scoring pair of identical length segments from two sequences Local alignment without gaps, similar to FASTA local region Expected distribution is known! Maximal Segment Pair sample calculation T G C A A T C G A T C G T C G T C C G T A T A C A : : : : : : : : : : : running sum A G C T C G T G A T C G T G G T G G G A T C G G T match = +1 mismatch = Potential MSP Potential MSP Gribskov 2.28
29 BLAST is based on Significant MSPs Scoring system Must have at least one positive score Expected score must be less than zero E = Σ f i s i Probability of an MSP scoring higher than S P(MSP>S) KNe -λs N = size of data, K and λ are constants Karlin, S., and Altschul, S.F., Proc.Natl.Acad.Sci. 87, , Gribskov 2.29
30 Normal Distribution Cumulative Cumulative Probability Probability Proba ability Gribskov 2.30
31 Extreme Value Distribution Cumulative 0.25 ive Probability Probabili Cumulat ity 0.2 Probability Run Length 0 Gribskov 2.31
32 BLAST Basic Idea Determine in advance the MSP score you need to be significant, S for example, choose S so that you will see fewer than 10 unrelated sequences in the database that score as high Look for matching words of length w that t score above a threshold, h T, such that MSPs of score S are unlikely to be missed. These are High-scoring Segment Pairs (HSPs) Gribskov 2.32
33 BLAST procedure Step 1: Compile list of high scoring words from query Step 2: Scan database for "hits" Step 3: Extend regions with 2 hits into MSPs Step 4: Dynamic programming alignment around MSPs sequence Gribskov 2.33
34 BLAST Step 1 - List of High Scoring Words Choose a significance level S Choose a word size, w, and cutoff, T, so that you are unlikely to miss MSPs with score S Make a table of all words in the "neighborhood" of the query (DNA sequences use all words) Typically 50 words for each residue Gribskov 2.34
35 BLAST Step 2 - Scan Database Scan only for words in neighborhood Use lookup tables (like FASTA) or finite automaton Keep data in memory to make it faster Gribskov 2.35
36 BLAST Step 3 - Extend Words to MSPs In BLAST2, a diagonal must have two word hits before extension to MSP is attempted. In principal, must examine diagonal until score drops to zero Shortcut, t only check until score drops by X T G C A A T C G A T C G T C G T C C G T A T A C A : : : : : : : : : : : A G C T C G T G A T C G T G G T G G G A T C G G T Potential MSP Potential MSP Gribskov 2.36
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST - Basic Local Alignment Search Tool
 Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Introduction to Computational Molecular Biology
 18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame
 1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
How to use KAIKObase Version 3.1.0
 How to use KAIKObase Version 3.1.0 Version3.1.0 29/Nov/2010 http://sgp2010.dna.affrc.go.jp/kaikobase/ Copyright National Institute of Agrobiological Sciences. All rights reserved. Outline 1. System overview
How to use KAIKObase Version 3.1.0 Version3.1.0 29/Nov/2010 http://sgp2010.dna.affrc.go.jp/kaikobase/ Copyright National Institute of Agrobiological Sciences. All rights reserved. Outline 1. System overview
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Adam M Phillippy Center for Bioinformatics and Computational Biology
 Adam M Phillippy Center for Bioinformatics and Computational Biology WGS sequencing shearing sequencing assembly WGS assembly Overlap reads identify reads with shared k-mers calculate edit distance Layout
Adam M Phillippy Center for Bioinformatics and Computational Biology WGS sequencing shearing sequencing assembly WGS assembly Overlap reads identify reads with shared k-mers calculate edit distance Layout
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Searching Sequence Databases
 Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
Exercise 2: Browser-Based Annotation and RNA-Seq Data
 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
BLAST. Basic Local Alignment Search Tool. Used to quickly compare a protein or DNA sequence to a database.
 BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
B L A S T! BLAST: Basic local alignment search tool. Copyright notice. February 6, Pairwise alignment: key points. Outline of tonight s lecture
 February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
Tutorial 4 BLAST Searching the CHO Genome
 Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
From Smith-Waterman to BLAST
 From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
Browser Exercises - I. Alignments and Comparative genomics
 Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
Meraculous De Novo Assembly of the Ariolimax dolichophallus Genome. Charles Cole, Jake Houser, Kyle McGovern, and Jennie Richardson
 Meraculous De Novo Assembly of the Ariolimax dolichophallus Genome Charles Cole, Jake Houser, Kyle McGovern, and Jennie Richardson Meraculous Assembler Published by the US Department of Energy Joint Genome
Meraculous De Novo Assembly of the Ariolimax dolichophallus Genome Charles Cole, Jake Houser, Kyle McGovern, and Jennie Richardson Meraculous Assembler Published by the US Department of Energy Joint Genome
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS
 FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
Chapter 4: Blast. Chaochun Wei Fall 2014
 Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Genome Assembly and De Novo RNAseq
 Genome Assembly and De Novo RNAseq BMI 7830 Kun Huang Department of Biomedical Informatics The Ohio State University Outline Problem formulation Hamiltonian path formulation Euler path and de Bruijin graph
Genome Assembly and De Novo RNAseq BMI 7830 Kun Huang Department of Biomedical Informatics The Ohio State University Outline Problem formulation Hamiltonian path formulation Euler path and de Bruijin graph
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:
 FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
Tutorial: How to use the Wheat TILLING database
 Tutorial: How to use the Wheat TILLING database Last Updated: 9/7/16 1. Visit http://dubcovskylab.ucdavis.edu/wheat_blast to go to the BLAST page or click on the Wheat BLAST button on the homepage. 2.
Tutorial: How to use the Wheat TILLING database Last Updated: 9/7/16 1. Visit http://dubcovskylab.ucdavis.edu/wheat_blast to go to the BLAST page or click on the Wheat BLAST button on the homepage. 2.
Scoring and heuristic methods for sequence alignment CG 17
 Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
Finishing Circular Assemblies. J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015
 Finishing Circular Assemblies J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Assembly Strategies de Bruijn graph Velvet, ABySS earlier, basic assemblers IDBA, SPAdes later, multi-k
Finishing Circular Assemblies J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Assembly Strategies de Bruijn graph Velvet, ABySS earlier, basic assemblers IDBA, SPAdes later, multi-k
Introduction to BLAST with Protein Sequences. Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2
 Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
FastA & the chaining problem
 FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
Pacific Symposium on Biocomputing 13: (2008) PASH 2.0: SCALEABLE SEQUENCE ANCHORING FOR NEXT-GENERATION SEQUENCING TECHNOLOGIES
 PASH 2.0: SCALEABLE SEQUENCE ANCHORING FOR NEXT-GENERATION SEQUENCING TECHNOLOGIES") PASH 2.0: SCALEABLE SEQUENCE ANCHORING FOR NEXT-GENERATION SEQUENCING TECHNOLOGIES CRISTIAN COARFA Human Genome Sequencing Center, Department of Molecular and Human Genetics, Baylor College of Medicine,
PASH 2.0: SCALEABLE SEQUENCE ANCHORING FOR NEXT-GENERATION SEQUENCING TECHNOLOGIES CRISTIAN COARFA Human Genome Sequencing Center, Department of Molecular and Human Genetics, Baylor College of Medicine,
Database Similarity Searching
 An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
Finding homologous sequences in databases
 Finding homologous sequences in databases There are multiple algorithms to search sequences databases BLAST (EMBL, NCBI, DDBJ, local) FASTA (EMBL, local) For protein only databases scan via Smith-Waterman
Finding homologous sequences in databases There are multiple algorithms to search sequences databases BLAST (EMBL, NCBI, DDBJ, local) FASTA (EMBL, local) For protein only databases scan via Smith-Waterman
Tutorial 1: Exploring the UCSC Genome Browser
 Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
AMOS Assembly Validation and Visualization
 AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland April 7, 2006 Outline AMOS Introduction Getting Data into AMOS AMOS
AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland April 7, 2006 Outline AMOS Introduction Getting Data into AMOS AMOS
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Sequence Alignment. GBIO0002 Archana Bhardwaj University of Liege
 Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
M 100 G 3000 M 3000 G 100. ii) iii)
 iii)") A) B) RefSeq 1 Other Alignments 180000 1 1 Simulation of Kim et al method Human Mouse Rat Fruitfly Nematode Best Alignment G estimate 1 80000 RefSeq 2 G estimate C) D) 0 350000 300000 250000 0 150000 Interpretation
A) B) RefSeq 1 Other Alignments 180000 1 1 Simulation of Kim et al method Human Mouse Rat Fruitfly Nematode Best Alignment G estimate 1 80000 RefSeq 2 G estimate C) D) 0 350000 300000 250000 0 150000 Interpretation
Jyoti Lakhani 1, Ajay Khunteta 2, Dharmesh Harwani *3 1 Poornima University, Jaipur & Maharaja Ganga Singh University, Bikaner, Rajasthan, India
 International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
Proteome Comparison: A fine-grained tool for comparative genomics
 Proteome Comparison: A fine-grained tool for comparative genomics In addition to the Protein Family Sorter that allows researchers to examine up to the protein families from up to 500 genomes at a time,
Proteome Comparison: A fine-grained tool for comparative genomics In addition to the Protein Family Sorter that allows researchers to examine up to the protein families from up to 500 genomes at a time,
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
INTRODUCTION TO CONSED
 INTRODUCTION TO CONSED OVERVIEW: Consed is a program that can be used to visually assemble and analyze sequence data. This introduction will take you through the basics of opening and operating within
INTRODUCTION TO CONSED OVERVIEW: Consed is a program that can be used to visually assemble and analyze sequence data. This introduction will take you through the basics of opening and operating within
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
L4: Blast: Alignment Scores etc.
 L4: Blast: Alignment Scores etc. Why is Blast Fast? Silly Question Prove or Disprove: There are two people in New York City with exactly the same number of hairs. Large database search Database (n) Query
L4: Blast: Alignment Scores etc. Why is Blast Fast? Silly Question Prove or Disprove: There are two people in New York City with exactly the same number of hairs. Large database search Database (n) Query
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching
 SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
Introduction to Bioinformatics Problem Set 3: Genome Sequencing
 Introduction to Bioinformatics Problem Set 3: Genome Sequencing 1. Assemble a sequence with your bare hands! You are trying to determine the DNA sequence of a very (very) small plasmids, which you estimate
Introduction to Bioinformatics Problem Set 3: Genome Sequencing 1. Assemble a sequence with your bare hands! You are trying to determine the DNA sequence of a very (very) small plasmids, which you estimate
Miniproject 1. Part 1 Due: 16 February. The coverage problem. Method. Why it is hard. Data. Task1
 Miniproject 1 Part 1 Due: 16 February The coverage problem given an assembled transcriptome (RNA) and a reference genome (DNA) 1. 2. what fraction (in bases) of the transcriptome sequences match to annotated
Miniproject 1 Part 1 Due: 16 February The coverage problem given an assembled transcriptome (RNA) and a reference genome (DNA) 1. 2. what fraction (in bases) of the transcriptome sequences match to annotated
PLNT4610 BIOINFORMATICS FINAL EXAMINATION
 9:00 to 11:00 Friday December 6, 2013 PLNT4610 BIOINFORMATICS FINAL EXAMINATION Answer any combination of questions totalling to exactly 100 points. The questions on the exam sheet total to 120 points.
9:00 to 11:00 Friday December 6, 2013 PLNT4610 BIOINFORMATICS FINAL EXAMINATION Answer any combination of questions totalling to exactly 100 points. The questions on the exam sheet total to 120 points.
Sequence Alignment Heuristics
 Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 13 G R A T I V. Iterative homology searching,
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
Single Pass, BLAST-like, Approximate String Matching on FPGAs*
 Single Pass, BLAST-like, Approximate String Matching on FPGAs* Martin Herbordt Josh Model Yongfeng Gu Bharat Sukhwani Tom VanCourt Computer Architecture and Automated Design Laboratory Department of Electrical
Single Pass, BLAST-like, Approximate String Matching on FPGAs* Martin Herbordt Josh Model Yongfeng Gu Bharat Sukhwani Tom VanCourt Computer Architecture and Automated Design Laboratory Department of Electrical
Example of repeats: ATGGTCTAGGTCCTAGTGGTC Motivation to find them: Genomic rearrangements are often associated with repeats Trace evolutionary
 Outline Hash Tables Repeat Finding Exact Pattern Matching Keyword Trees Suffix Trees Heuristic Similarity Search Algorithms Approximate String Matching Filtration Comparing a Sequence Against a Database
Outline Hash Tables Repeat Finding Exact Pattern Matching Keyword Trees Suffix Trees Heuristic Similarity Search Algorithms Approximate String Matching Filtration Comparing a Sequence Against a Database
Omega: an Overlap-graph de novo Assembler for Metagenomics
 Omega: an Overlap-graph de novo Assembler for Metagenomics B a h l e l H a i d e r, Ta e - H y u k A h n, B r i a n B u s h n e l l, J u a n j u a n C h a i, A l e x C o p e l a n d, C h o n g l e Pa n
Omega: an Overlap-graph de novo Assembler for Metagenomics B a h l e l H a i d e r, Ta e - H y u k A h n, B r i a n B u s h n e l l, J u a n j u a n C h a i, A l e x C o p e l a n d, C h o n g l e Pa n
Computational models for bionformatics
 Computational models for bionformatics De-novo assembly and alignment-free measures Michele Schimd Department of Information Engineering July 8th, 2015 Michele Schimd (DEI) PostDoc @ DEI July 8th, 2015
Computational models for bionformatics De-novo assembly and alignment-free measures Michele Schimd Department of Information Engineering July 8th, 2015 Michele Schimd (DEI) PostDoc @ DEI July 8th, 2015
MacVector for Mac OS X. The online updater for this release is MB in size
 MacVector 17.0.3 for Mac OS X The online updater for this release is 143.5 MB in size You must be running MacVector 15.5.4 or later for this updater to work! System Requirements MacVector 17.0 is supported
MacVector 17.0.3 for Mac OS X The online updater for this release is 143.5 MB in size You must be running MacVector 15.5.4 or later for this updater to work! System Requirements MacVector 17.0 is supported
CodonCode Aligner User Manual
 CodonCode Aligner User Manual CodonCode Aligner User Manual Table of Contents About CodonCode Aligner...1 System Requirements...1 Licenses...1 Licenses for CodonCode Aligner...3 Demo Mode...3 Time-limited
CodonCode Aligner User Manual CodonCode Aligner User Manual Table of Contents About CodonCode Aligner...1 System Requirements...1 Licenses...1 Licenses for CodonCode Aligner...3 Demo Mode...3 Time-limited
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J.
 BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J. Buhler Prerequisites: BLAST Exercise: Detecting and Interpreting
BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J. Buhler Prerequisites: BLAST Exercise: Detecting and Interpreting
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL
 QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
Genomic Finishing & Consed
 Genomic Finishing & Consed SEA stages of genomic analysis Draft vs Finished Draft Sequence Single sequencing approach Limited human intervention Cheap, Fast Finished sequence Multiple approaches Human
Genomic Finishing & Consed SEA stages of genomic analysis Draft vs Finished Draft Sequence Single sequencing approach Limited human intervention Cheap, Fast Finished sequence Multiple approaches Human
ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS
 ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS Ivan Vogel Doctoral Degree Programme (1), FIT BUT E-mail: xvogel01@stud.fit.vutbr.cz Supervised by: Jaroslav Zendulka E-mail: zendulka@fit.vutbr.cz
ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS Ivan Vogel Doctoral Degree Programme (1), FIT BUT E-mail: xvogel01@stud.fit.vutbr.cz Supervised by: Jaroslav Zendulka E-mail: zendulka@fit.vutbr.cz
Alignments BLAST, BLAT
 Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Two Examples of Datanomic. David Du Digital Technology Center Intelligent Storage Consortium University of Minnesota
 Two Examples of Datanomic David Du Digital Technology Center Intelligent Storage Consortium University of Minnesota Datanomic Computing (Autonomic Storage) System behavior driven by characteristics of
Two Examples of Datanomic David Du Digital Technology Center Intelligent Storage Consortium University of Minnesota Datanomic Computing (Autonomic Storage) System behavior driven by characteristics of
Tour Guide for Windows and Macintosh
 Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
A Design of a Hybrid System for DNA Sequence Alignment
 IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
17 ½ Weeks in Leipzig, Saxonia. Andreas Gruber Institute for Theoretical Chemistry University of Vienna
 17 ½ Weeks in Leipzig, Saxonia Andreas Gruber Institute for Theoretical Chemistry University of Vienna START Leipzig, 1. 6. 2009 Idea? RNAz FINISH Vienna, 1. 10. 2009 START Leipzig, 1. 6. 2009 Idea? RNAz
17 ½ Weeks in Leipzig, Saxonia Andreas Gruber Institute for Theoretical Chemistry University of Vienna START Leipzig, 1. 6. 2009 Idea? RNAz FINISH Vienna, 1. 10. 2009 START Leipzig, 1. 6. 2009 Idea? RNAz
Important Example: Gene Sequence Matching. Corrigiendum. Central Dogma of Modern Biology. Genetics. How Nucleotides code for Amino Acids
 Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Variant calling using SAMtools
 Variant calling using SAMtools Calling variants - a trivial use of an Interactive Session We are going to conduct the variant calling exercises in an interactive idev session just so you can get a feel
Variant calling using SAMtools Calling variants - a trivial use of an Interactive Session We are going to conduct the variant calling exercises in an interactive idev session just so you can get a feel
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
PLNT4610 BIOINFORMATICS FINAL EXAMINATION
 PLNT4610 BIOINFORMATICS FINAL EXAMINATION 18:00 to 20:00 Thursday December 13, 2012 Answer any combination of questions totalling to exactly 100 points. The questions on the exam sheet total to 120 points.
PLNT4610 BIOINFORMATICS FINAL EXAMINATION 18:00 to 20:00 Thursday December 13, 2012 Answer any combination of questions totalling to exactly 100 points. The questions on the exam sheet total to 120 points.
) I R L Press Limited, Oxford, England. The protein identification resource (PIR)
 I R L Press Limited, Oxford, England. The protein identification resource (PIR)") Volume 14 Number 1 Volume 1986 Nucleic Acids Research 14 Number 1986 Nucleic Acids Research The protein identification resource (PIR) David G.George, Winona C.Barker and Lois T.Hunt National Biomedical
Volume 14 Number 1 Volume 1986 Nucleic Acids Research 14 Number 1986 Nucleic Acids Research The protein identification resource (PIR) David G.George, Winona C.Barker and Lois T.Hunt National Biomedical
(for more info see:
 Genome assembly (for more info see: http://www.cbcb.umd.edu/research/assembly_primer.shtml) Introduction Sequencing technologies can only "read" short fragments from a genome. Reconstructing the entire
Genome assembly (for more info see: http://www.cbcb.umd.edu/research/assembly_primer.shtml) Introduction Sequencing technologies can only "read" short fragments from a genome. Reconstructing the entire