Aligners. J Fass 21 June 2017
|
|
|
- Ira Rogers
- 6 years ago
- Views:
Transcription
1 Aligners J Fass 21 June 2017
2 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners
3 Definitions Alignment: Somebody plagiarized parts of my document; where did they copy paragraphs from and where did each of the words come from?
4 Definitions Mapping: Somebody plagiarized parts of my document; where did they copy paragraphs from and where did each of the words come from?

5 Why align (or map)? Gene X align / map Gene X To infer abundance:

6 Why align (or map)? Individual A Individual B To measure variation
7 Why align (or map)?
8 More Definitions: Global and local Global aligners try to align all provided sequence, end to end, both query and subject / target E.g. Aligning two Salmonella genomes Aligning human and gorilla orthologous coding regions
9 Global and local Local aligners try to find hits or chains of hits within each provided sequence E.g. Finding mitochondrial splinters in nuclear chromosomes Finding genes that share a domain with a gene of interest
10 Glocal? Short read aligners generally assume that the whole read came from somewhere within the target (reference) sequence so, global with respect to the read, and local with respect to the reference.
11 Short Read (Non-splicing) Aligners Li, H and Homer, N (2010) Briefings in Bioinformatics 11:473 A survey of sequence alignment algorithms for next-generation sequencing These two were fastest, at ~7 Gbp (vs human) per CPU day HiSeq 2500 generated Gbp per day (at the time) (Fall 12-13) Gbp per day (Summer 16) 600 Gbp per day (Summer 17) 1-3 Tbp per day
12 Burrows-Wheeler Aligners Burrows-Wheeler Transform used in bzip2 file compression tool; FM-index (Ferragina & Manzini) allow efficient finding of substring matches within compressed text algorithm is sub-linear with respect to time and storage space required for a certain set of input data (reference 'ome, essentially). Reduced memory footprint, faster execution.
13 BWA BWA is a fast gapped aligner. Long read aligners (bwasw and mem) also fast, and can perform well for 454, Ion Torrent, Sanger, and PacBio reads. BWA is actively maintained and has a strong user community. bio-bwa.sourceforge.net bwa aln (BWA backtrack ) for reads < 70 bp bwa bwasw bwa mem (seeds with maximal exact matches, extends via Smith-Waterman)
14 Bowtie (now Bowtie 2) comparable to BWA. Bowtie is part of a suite of tools (Bowtie, Tophat, Cufflinks, CummeRbund) that address RNAseq experiments. Written by same folks as Tophat so, full compatibility.
15 Tophat2 Aligns full reads to genome, to determine coverage islands. Creates simulated spliced exons based on these islands, then aligns remaining short reads to the simulated cdna (to find reads crossing splice junctions).
16 STAR Spliced Transcripts Aligned to a Reference Aligns short reads and full length cdna Similar to BWA MEM algorithm, but searches uncompressed version (suffix array) of genome (faster, but more RAM required!). Claims better sensitivity and specificity than previous short read aligners, and 50x speed vs TopHat2. Requires ~27 GB RAM for human genome!
17 HISAT2 Improved replacement for Tophat2, multiple high level and low level indexes of compressed sequence (similar to graph-based assemblers using De Bruijn graphs to represent overlapping k-mers). Graph structure can represent multiple sequence paths, i.e. a population of references, allowing rapid genotyping of samples. HISAT2 is currently probably the best combination, in a full aligner, of speed (faster than STAR) and memory (~4 GB for human genome). Kim, Langmead, Salzberg (2015) Nature Methods 12:357
 and bootstrapping to determine most likely transcript and abundance uncertainty. Bray Pachter (2016) Nat Biotech 34:525 See sleuth for DGE analysis.")
18 Kallisto A pseudoaligner (mapper? see Pachter blog post) that compares read k-mers (overlapping subsequences) to a transcriptome de Bruijn graph (T-DBG) to find transcripts compatible with the read. Also uses expectation maximization (EM) and bootstrapping to determine most likely transcript and abundance uncertainty. Bray Pachter (2016) Nat Biotech 34:525 See sleuth for DGE analysis.
19 Salmon Supersedes Sailfish, an older k-mer based transcript analysis tool. Performs quasi-mapping to a set of transcripts (not genome), similar in methods to kallisto. Also performs bias correction for multiple modes of bias (sequence, position) to more accurately determine abundance. Patro, Duggard, Kingsford (2015) biorxiv
20 General Alignment Parameters / Concepts

21 General Alignment Parameters / Concepts
22 General Alignment Parameters / Concepts Clipping: 134M 16S(H) Splicing: 32M27N59M

23 General Alignment Parameters / Concepts
24 General Alignment Parameters / Concepts Multimappers: Reads that align equally well to more than one reference location. Generally, multimappers are discounted in variant detection, and are often discounted in counting applications (like RNA-Seq would cancel out anyway). Note: multimapper rescue in some algorithms (RSEM, Express?). Duplicates: Reads or read pairs arising from the same original library fragment, either during library preparation (PCR duplicates) or colony formation (optical duplicates; not an issue anymore). Generally, duplicates can only be detected reliably with paired-end sequencing. If PE, they re discounted in variant detection, and discounted in counting applications (like RNA-Seq).
25 General Alignment Parameters / Concepts
26
, IGB, GenomeView, SAMscope.")
27 Alignment Viewers IGV (Integrated Genomics Viewer) BAMview, tview (in SAMtools), IGB, GenomeView, SAMscope... UCSC Genome Browser, GBrowse
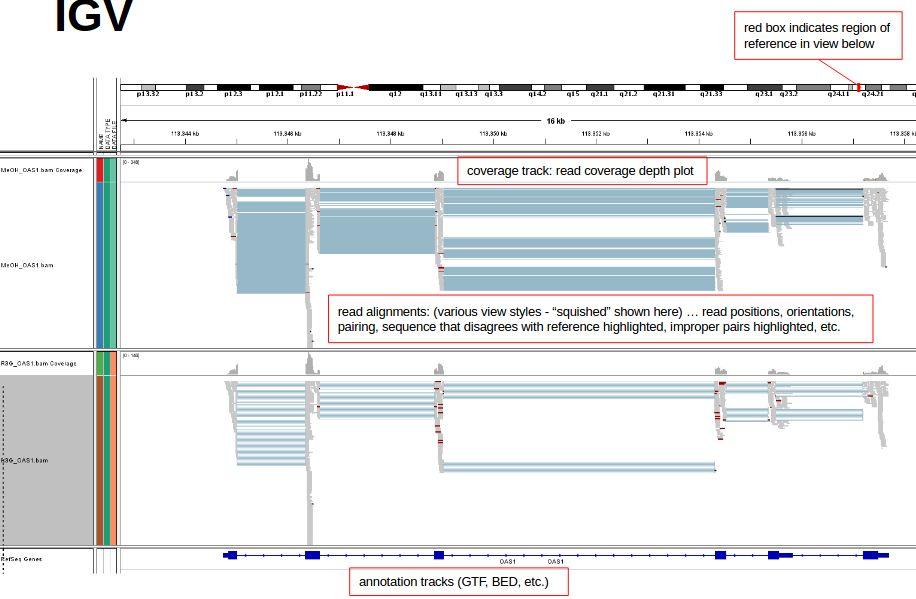
28 IGV

29 IGV
30 IGV More on IGV s interface, file formats, and display can be found here: More on interpreting and customizing IGV s display can be found here:
Aligners. J Fass 23 August 2017
 Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Introduction to Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014
 Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014 Deciphering the information contained in DNA sequences began decades ago since the time of Sanger sequencing.
Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014 Deciphering the information contained in DNA sequences began decades ago since the time of Sanger sequencing.
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Read Mapping. Slides by Carl Kingsford
 Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Aligning reads: tools and theory
 Aligning reads: tools and theory Genome Sequence read :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-14pos :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg chrx: 152139280 152139290 152139300
Aligning reads: tools and theory Genome Sequence read :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-14pos :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg chrx: 152139280 152139290 152139300
Long Read RNA-seq Mapper
 UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
Read Mapping and Assembly
 Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Scalable RNA Sequencing on Clusters of Multicore Processors
 JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
SAM and VCF formats. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 SAM and VCF formats UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 File Format: SAM / BAM / CRAM! NEW http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
SAM and VCF formats UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 File Format: SAM / BAM / CRAM! NEW http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
KisSplice. Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data. 29th may 2013
 Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)
Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
Sequence mapping and assembly. Alistair Ward - Boston College
 Sequence mapping and assembly Alistair Ward - Boston College Sequenced a genome? Fragmented a genome -> DNA library PCR amplification Sequence reads (ends of DNA fragment for mate pairs) We no longer have
Sequence mapping and assembly Alistair Ward - Boston College Sequenced a genome? Fragmented a genome -> DNA library PCR amplification Sequence reads (ends of DNA fragment for mate pairs) We no longer have
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Accelerating Genomic Sequence Alignment Workload with Scalable Vector Architecture
 Accelerating Genomic Sequence Alignment Workload with Scalable Vector Architecture Dong-hyeon Park, Jon Beaumont, Trevor Mudge University of Michigan, Ann Arbor Genomics Past Weeks ~$3 billion Human Genome
Accelerating Genomic Sequence Alignment Workload with Scalable Vector Architecture Dong-hyeon Park, Jon Beaumont, Trevor Mudge University of Michigan, Ann Arbor Genomics Past Weeks ~$3 billion Human Genome
Data Processing and Analysis in Systems Medicine. Milena Kraus Data Management for Digital Health Summer 2017
 Milena Kraus Digital Health Summer Agenda Real-world Use Cases Oncology Nephrology Heart Insufficiency Additional Topics Data Management & Foundations Biology Recap Data Sources Data Formats Business Processes
Milena Kraus Digital Health Summer Agenda Real-world Use Cases Oncology Nephrology Heart Insufficiency Additional Topics Data Management & Foundations Biology Recap Data Sources Data Formats Business Processes
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching
 SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
A review of RNA-Seq normalization methods
 A review of RNA-Seq normalization methods This post covers the units used in RNA-Seq that are, unfortunately, often misused and misunderstood I ll try to clear up a bit of the confusion here The first
A review of RNA-Seq normalization methods This post covers the units used in RNA-Seq that are, unfortunately, often misused and misunderstood I ll try to clear up a bit of the confusion here The first
Next generation sequencing: assembly by mapping reads. Laurent Falquet, Vital-IT Helsinki, June 3, 2010
 Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Short Read Alignment Algorithms
 Short Read Alignment Algorithms Raluca Gordân Department of Biostatistics and Bioinformatics Department of Computer Science Department of Molecular Genetics and Microbiology Center for Genomic and Computational
Short Read Alignment Algorithms Raluca Gordân Department of Biostatistics and Bioinformatics Department of Computer Science Department of Molecular Genetics and Microbiology Center for Genomic and Computational
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat a.tgz. Software:
 A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
Gene Expression Data Analysis. Qin Ma, Ph.D. December 10, 2017
 1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
Read mapping with BWA and BOWTIE
 Read mapping with BWA and BOWTIE Before We Start In order to save a lot of typing, and to allow us some flexibility in designing these courses, we will establish a UNIX shell variable BASE to point to
Read mapping with BWA and BOWTIE Before We Start In order to save a lot of typing, and to allow us some flexibility in designing these courses, we will establish a UNIX shell variable BASE to point to
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units
 GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Data Preprocessing. Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
A Fast Read Alignment Method based on Seed-and-Vote For Next GenerationSequencing
 A Fast Read Alignment Method based on Seed-and-Vote For Next GenerationSequencing Song Liu 1,2, Yi Wang 3, Fei Wang 1,2 * 1 Shanghai Key Lab of Intelligent Information Processing, Shanghai, China. 2 School
A Fast Read Alignment Method based on Seed-and-Vote For Next GenerationSequencing Song Liu 1,2, Yi Wang 3, Fei Wang 1,2 * 1 Shanghai Key Lab of Intelligent Information Processing, Shanghai, China. 2 School
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
version /1/2011 Source code Linux x86_64 binary Mac OS X x86_64 binary
 Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
Quark enables semi-reference-based compression of RNA-seq data
 Bioinformatics, 33(21), 2017, 3380 3386 doi: 10.1093/bioinformatics/btx428 Advance Access Publication Date: 3 July 2017 Original Paper Sequence analysis Quark enables semi-reference-based compression of
Bioinformatics, 33(21), 2017, 3380 3386 doi: 10.1093/bioinformatics/btx428 Advance Access Publication Date: 3 July 2017 Original Paper Sequence analysis Quark enables semi-reference-based compression of
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Our data for today is a small subset of Saimaa ringed seal RNA sequencing data (RNA_seq_reads.fasta). Let s first see how many reads are there:
. Let s first see how many reads are there:") Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Gene expression estimation
 Gene expression estimation") mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
Services Performed. The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples.
 Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Goal: Learn how to use various tool to extract information from RNAseq reads. 4.1 Mapping RNAseq Reads to a Genome Assembly
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Mapping Reads to Reference Genome
 Mapping Reads to Reference Genome DNA carries genetic information DNA is a double helix of two complementary strands formed by four nucleotides (bases): Adenine, Cytosine, Guanine and Thymine 2 of 31 Gene
Mapping Reads to Reference Genome DNA carries genetic information DNA is a double helix of two complementary strands formed by four nucleotides (bases): Adenine, Cytosine, Guanine and Thymine 2 of 31 Gene
A natural encoding of genetic variation in a Burrows-Wheeler Transform to enable mapping and genome inference
 Maciuca et al. RESEARCH A natural encoding of genetic variation in a Burrows-Wheeler Transform to enable mapping and genome inference Sorina Maciuca 1, Carlos del Ojo Elias 2, Gil McVean 1 and Zamin Iqbal
Maciuca et al. RESEARCH A natural encoding of genetic variation in a Burrows-Wheeler Transform to enable mapping and genome inference Sorina Maciuca 1, Carlos del Ojo Elias 2, Gil McVean 1 and Zamin Iqbal
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
The Burrows-Wheeler Transform and Bioinformatics. J. Matthew Holt April 1st, 2015
 The Burrows-Wheeler Transform and Bioinformatics J. Matthew Holt April 1st, 2015 Outline Recall Suffix Arrays The Burrows-Wheeler Transform The FM-index Pattern Matching Multi-string BWTs Merge Algorithms
The Burrows-Wheeler Transform and Bioinformatics J. Matthew Holt April 1st, 2015 Outline Recall Suffix Arrays The Burrows-Wheeler Transform The FM-index Pattern Matching Multi-string BWTs Merge Algorithms
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Bioinformatics for High-throughput Sequencing
 Bioinformatics for High-throughput Sequencing An Overview Simon Anders EBI is an Outstation of the European Molecular Biology Laboratory. Overview In recent years, new sequencing schemes, also called high-throughput
Bioinformatics for High-throughput Sequencing An Overview Simon Anders EBI is an Outstation of the European Molecular Biology Laboratory. Overview In recent years, new sequencing schemes, also called high-throughput
Shotgun sequencing. Coverage is simply the average number of reads that overlap each true base in genome.
 Shotgun sequencing Genome (unknown) Reads (randomly chosen; have errors) Coverage is simply the average number of reads that overlap each true base in genome. Here, the coverage is ~10 just draw a line
Shotgun sequencing Genome (unknown) Reads (randomly chosen; have errors) Coverage is simply the average number of reads that overlap each true base in genome. Here, the coverage is ~10 just draw a line
Data Preprocessing : Next Generation Sequencing analysis CBS - DTU Next Generation Sequencing Analysis
 Data Preprocessing 27626: Next Generation Sequencing analysis CBS - DTU Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing 27626: Next Generation Sequencing analysis CBS - DTU Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
The Burrows-Wheeler Transform and Bioinformatics. J. Matthew Holt
 The Burrows-Wheeler Transform and Bioinformatics J. Matthew Holt holtjma@cs.unc.edu Last Class - Multiple Pattern Matching Problem m - length of text d - max length of pattern x - number of patterns Method
The Burrows-Wheeler Transform and Bioinformatics J. Matthew Holt holtjma@cs.unc.edu Last Class - Multiple Pattern Matching Problem m - length of text d - max length of pattern x - number of patterns Method
11/8/2017 Trinity De novo Transcriptome Assembly Workshop trinityrnaseq/rnaseq_trinity_tuxedo_workshop Wiki GitHub
 trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
RNAseq analysis: SNP calling. BTI bioinformatics course, spring 2013
 RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
File Formats: SAM, BAM, and CRAM. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide
 RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
Finding the appropriate method, with a special focus on: Mapping and alignment. Philip Clausen
 Finding the appropriate method, with a special focus on: Mapping and alignment Philip Clausen Background Most people choose their methods based on popularity and history, not by reasoning and research.
Finding the appropriate method, with a special focus on: Mapping and alignment Philip Clausen Background Most people choose their methods based on popularity and history, not by reasoning and research.
SpliceTAPyR - An efficient method for transcriptome alignment
 International Journal of Foundations of Computer Science c World Scientific Publishing Company SpliceTAPyR - An efficient method for transcriptome alignment Andreia Sofia Teixeira IDSS Lab, INESC-ID /
International Journal of Foundations of Computer Science c World Scientific Publishing Company SpliceTAPyR - An efficient method for transcriptome alignment Andreia Sofia Teixeira IDSS Lab, INESC-ID /
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Integrated Genome browser (IGB) installation
 installation") Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
Variation among genomes
 Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
Ballgown. flexible RNA-seq differential expression analysis. Alyssa Frazee Johns Hopkins
 Ballgown flexible RNA-seq differential expression analysis Alyssa Frazee Johns Hopkins Biostatistics @acfrazee RNA-seq data Reads (50-100 bases) Transcripts (RNA) Genome (DNA) [use tool of your choice]
Ballgown flexible RNA-seq differential expression analysis Alyssa Frazee Johns Hopkins Biostatistics @acfrazee RNA-seq data Reads (50-100 bases) Transcripts (RNA) Genome (DNA) [use tool of your choice]
RNA-seq. Read mapping and Quantification. Genomics: Lecture #12. Institut für Medizinische Genetik und Humangenetik Charité Universitätsmedizin Berlin
 (1) Read and Quantification Institut für Medizinische Genetik und Humangenetik Charité Universitätsmedizin Berlin Genomics: Lecture #12 Today (1) Gene Expression Previous gold standard: Basic protocol
(1) Read and Quantification Institut für Medizinische Genetik und Humangenetik Charité Universitätsmedizin Berlin Genomics: Lecture #12 Today (1) Gene Expression Previous gold standard: Basic protocol
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
USING BRAT-BW Table 1. Feature comparison of BRAT-bw, BRAT-large, Bismark and BS Seeker (as of on March, 2012)
") USING BRAT-BW-2.0.1 BRAT-bw is a tool for BS-seq reads mapping, i.e. mapping of bisulfite-treated sequenced reads. BRAT-bw is a part of BRAT s suit. Therefore, input and output formats for BRAT-bw are
USING BRAT-BW-2.0.1 BRAT-bw is a tool for BS-seq reads mapping, i.e. mapping of bisulfite-treated sequenced reads. BRAT-bw is a part of BRAT s suit. Therefore, input and output formats for BRAT-bw are
Concurrent and Accurate RNA Sequencing on Multicore Platforms
 Technical Report ICC 2013-03-01 Concurrent and Accurate RNA Sequencing on Multicore Platforms Héctor Martínez *, Joaquín Tárraga, Ignacio Medina, Sergio Barrachina *, Maribel Castillo *, Joaquín Dopazo,
Technical Report ICC 2013-03-01 Concurrent and Accurate RNA Sequencing on Multicore Platforms Héctor Martínez *, Joaquín Tárraga, Ignacio Medina, Sergio Barrachina *, Maribel Castillo *, Joaquín Dopazo,
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1
 EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
Sequence Preprocessing: A perspective
 Sequence Preprocessing: A perspective Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu Why Preprocess reads We have found that aggressively cleaning and processing
Sequence Preprocessing: A perspective Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu Why Preprocess reads We have found that aggressively cleaning and processing
Genome Assembly Using de Bruijn Graphs. Biostatistics 666
 Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Browser Exercises - I. Alignments and Comparative genomics
 Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
From the Schnable Lab:
 From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
A Tutorial: Genome- based RNA- Seq Analysis Using the TUXEDO Package
 A Tutorial: Genome- based RNA- Seq Analysis Using the TUXEDO Package The following data and software resources are required for following the tutorial. Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
A Tutorial: Genome- based RNA- Seq Analysis Using the TUXEDO Package The following data and software resources are required for following the tutorial. Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
de Bruijn graphs for sequencing data
 de Bruijn graphs for sequencing data Rayan Chikhi CNRS Bonsai team, CRIStAL/INRIA, Univ. Lille 1 SMPGD 2016 1 MOTIVATION - de Bruijn graphs are instrumental for reference-free sequencing data analysis:
de Bruijn graphs for sequencing data Rayan Chikhi CNRS Bonsai team, CRIStAL/INRIA, Univ. Lille 1 SMPGD 2016 1 MOTIVATION - de Bruijn graphs are instrumental for reference-free sequencing data analysis:
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Darwin-WGA. A Co-processor Provides Increased Sensitivity in Whole Genome Alignments with High Speedup
 Darwin-WGA A Co-processor Provides Increased Sensitivity in Whole Genome Alignments with High Speedup Yatish Turakhia*, Sneha D. Goenka*, Prof. Gill Bejerano, Prof. William J. Dally * Equal contribution
Darwin-WGA A Co-processor Provides Increased Sensitivity in Whole Genome Alignments with High Speedup Yatish Turakhia*, Sneha D. Goenka*, Prof. Gill Bejerano, Prof. William J. Dally * Equal contribution
Using Galaxy: RNA-seq
 Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Tiling Assembly for Annotation-independent Novel Gene Discovery
 Tiling Assembly for Annotation-independent Novel Gene Discovery By Jennifer Lopez and Kenneth Watanabe Last edited on September 7, 2015 by Kenneth Watanabe The following procedure explains how to run the
Tiling Assembly for Annotation-independent Novel Gene Discovery By Jennifer Lopez and Kenneth Watanabe Last edited on September 7, 2015 by Kenneth Watanabe The following procedure explains how to run the
On enhancing variation detection through pan-genome indexing
 Standard approach...t......t......t......acgatgctagtgcatgt......t......t......t... reference genome Variation graph reference SNP: A->T...ACGATGCTTGTGCATGT donor genome Can we boost variation detection
Standard approach...t......t......t......acgatgctagtgcatgt......t......t......t... reference genome Variation graph reference SNP: A->T...ACGATGCTTGTGCATGT donor genome Can we boost variation detection
Resequencing and Mapping. Andreas Gisel Inernational Institute of Tropical Agriculture (IITA) Ibadan, Nigeria
 Ibadan, Nigeria") Resequencing and Mapping Andreas Gisel Inernational Institute of Tropical Agriculture (IITA) Ibadan, Nigeria The Principle of Mapping reads good, ood_, d_mo, morn, orni, ning, ing_, g_be, beau, auti, utif,
Resequencing and Mapping Andreas Gisel Inernational Institute of Tropical Agriculture (IITA) Ibadan, Nigeria The Principle of Mapping reads good, ood_, d_mo, morn, orni, ning, ing_, g_be, beau, auti, utif,
AMAS: optimizing the partition and filtration of adaptive seeds to speed up read mapping
 AMAS: optimizing the partition and filtration of adaptive seeds to speed up read mapping Ngoc Hieu Tran 1, * Email: nhtran@ntu.edu.sg Xin Chen 1 Email: chenxin@ntu.edu.sg 1 School of Physical and Mathematical
AMAS: optimizing the partition and filtration of adaptive seeds to speed up read mapping Ngoc Hieu Tran 1, * Email: nhtran@ntu.edu.sg Xin Chen 1 Email: chenxin@ntu.edu.sg 1 School of Physical and Mathematical
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq
 TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018)
") Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Exercise 2: Browser-Based Annotation and RNA-Seq Data
 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola