Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
|
|
|
- Annabel Porter
- 6 years ago
- Views:
Transcription
1 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility

2 RNA-seq workflow I Hypothesis (a.k.a. the research question) Differentially expressed genes across several conditions of an experiment Simple two conditions: Wild type vs. gene knockout mouse Healthy person vs. cancer patient Control vs. treatment with drug Complexity can increase arbitrarily: Many conditions, confounding factors, time course experiments, etc.

3 RNA-seq workflow I Experimental design Important to ensure (statistical) validity of results Depends on the hypothesis: Cell cultures or animals/patients? Phenotypic effect mild or severe? Inclusion of non-coding RNA?... Affects choice of protocols for culturing, RNA extraction, sample preparation, sequencing, bioinformatics and esp. number of replicates per condition! Involve statistician/bioinformatician from the beginning!
4 RNA-seq workflow I Sequencing processing Post-processing of intensity values basecalling: convert sequence of intensities to nucleotide sequences ( reads ) demultiplexing: assign reads to samples based on their adapter sequences ( barcodes ) Sample-specific sequence read files Fragments can be sequenced from one or both ends unpaired / single-end vs. paired-end RNA-seq often run with single-end

")
5 RNA-seq workflow II FASTQ the sequencing read file format Raw reads from sample-specific fragments Per-base quality information (Phred score 33) biocluster.ucr.edu
6 RNA-seq workflow II FASTQ processing Steps towards identifying differential expression of genes between samples: 1) Quality assessment of raw reads 2) Alignment of reads to the genome 3) Quantification of gene expression QC of Raw Reads Read Alignment How can I do that on my own? Quantification

7 Galaxy Open source, web-based platform for data intensive biomedical research developed at Penn State and Johns Hopkins University Many (NGS) bioinformatics tools available as plug-ins Container-based server runs in a container that can be installed and customized on other systems many instances of Galaxy running worldwide User works on histories of data and processes, data can be shared with other users Galaxy@GWDG:

8 Galaxy practical I Open and login with your GWDG/course account
9 Galaxy practical I Uploading data into Galaxy a sandbox example: Go to Click Downloads, then Download data via FTP Click on GTF for Human Gene sets Download Homo_sapiens.GRCh38.90.gtf.gz to your PC Go back to Galaxy Click Get Data, then Upload File from your computer Choose local file from your PC (check Download folder) If successful, close the window Optional: rename history (click on unnamed history )

10 You should see this: Your history should look like this:

11 Galaxy practical I Uploading data may be time-consuming Galaxy allows importing data from public repositories and sharing data with other users We shared a data set from a published study: Published January 2017
12 Galaxy practical I Shared Data Data Libraries RNA-Seq_MolBio_Lecture Raw Data 3 control condition samples ( GFP... ), 3 overexpression samples ( PCDH7... ) Click any of the files to inspect data Add all files to your history; several options: Individually open files and click to History (slow) Mark files in folder view and click to History (fast) Mark whole folder and click to History (fast) Import into existing history, go to Main menu and click the eye symbol for one of the samples

13 You should see this:

14 Zoom in to see FastQ file features read nucleotide sequence base quality information read length
15 RNA-seq workflow II essential questions about quality control How many reads should I have? >=25 million reads required for representative transcriptome profile of model organisms such as human and mouse PCR introduces many (uninformative) duplicates How good are the reads? Assess signal-to-noise ratio of sequencing Determine proportion of ambigous bases ( N ) Identify fraction of adapters, contamination, etc.

16 RNA-seq workflow II Phred scores reflecting on basecall accuracy How good are the bases/reads? Phred scale: logarithmic scale of basecall accuracy Common threshold for good quality Phred Quality Score Probability of Incorrect Basecall Basecall accuracy 10 1 in 10 90% 20 1 in % 30 1 in % 40 1 in % 50 1 in %
17 RNA-seq workflow II Quality control indices Further quality indices: Distribution of nucleotide frequencies across the sequences GC content per sequence Fraction of N Length distribution of sequences Sequence duplication level Amount of overrepresented sequences and short (6-8 bp) stretches of nucleotides ( k-mers ) Adapter content trimming may be required

18 RNA-seq workflow II FastQC: A quality control tool for high throughput sequence data Systematically assess quality for NGS samples in Galaxy FastQC Open source tool Runs on all platforms Assess various quality parameters including contamination by adapters Allows to provide contamination sequences by user Generates intuitively interpretable output and visualization

19 RNA-seq workflow II FastQC per base quality scores


20 Galaxy practical II Quality control with FastQC General Sequencing Quality Control FastQC and read the description Click Multiple datasets and select all FASTQ files from your history Click Execute
21 Galaxy practical II Quality control with FastQC Execution calls several instances of the FastQC program, which are scheduled by the server execution time depends on file size, number of files, number of users and server load After a few minutes you should see FastQC results in your history (hit refresh symbol if not) As soon as any job is finished you can inspect the results choose Webpage, then eye symbol Scroll through the Webpage we are here to answer your questions! FastQC RawData contains detailed reports

22 RNA-seq workflow III Short read alignment Goal: determine the origin of sequenced reads w.r.t. the genome
23 RNA-seq workflow III Short read alignment Sequence alignment: Re-arrangement of two or more biological sequences to identify corresponding nucleotides/amino acids Example: sequence 1: sequence 2: ACATCGA ACTAGCTA possible alignment: ACATCG--A AC-TAGCTA
24 RNA-seq workflow III Short read alignment Terminology: match: two residues in a position match mismatch: residue is substituted by different residue gap: residue(s) is/are inserted or deleted match insertion ACATCG--A AC-TAGCTA deletion mismatch
25 RNA-seq workflow III Short read alignment Quality of an aligment: alignment score: sum of quality of position matches Example: position scores: match=+1, mismatch=-1, gap=-1 possibility 1: possibility 2: A C A T C G - - A A C - T A G C T A A C A T C - G - - A A C - T - A G C T A score: 5*1 + 4*(-1)=1 score: 5*1 + 5*(-1)=0
sequence(s) within longer")
26 RNA-seq workflow III Short read alignment Global vs local aligment: Global: align sequences end-to-end Local: find optimal placement of (sub)sequence(s) within longer sequence
27 RNA-seq workflow III Short read alignment Application of sequence alignment: Homology detection: identify best match of a sequence to many sequences in a database e.g. NCBI BLAST Identify conserved sites via multiple alignments of related protein sequences e.g. EMBL-EBI Clustal Omega Short read alignment ( mapping ): Identify origin of a sequence w.r.t. a genomic reference sequence e.g. Bowtie, BWA, TopHat, STAR, HiSAT,...
28 RNA-seq workflow III Short read alignment Reference sequence: complement of DNA sequences (genome) or mrna sequences (transcriptome) from an organism usually provided as (multi-)fasta file containing one sequence per chromosome/transcript completeness and complexity depends on organism's genome project advance: Organism Assembly Length (Mb) Chromosomes Human (Homo sapiens) GRCh38.p chromosomes, 2 sex chromosomes and nonnuclear mitochondrial DNA African clawed frog (Xenopus laevis) Xenopus_laevi s_v chromosomes, non-nuclear mitochondrial DNA Genes
29 RNA-seq workflow III Short read alignment Transcriptome sizes are substantially smaller, e.g. human transcriptome: 20,338 coding genes 22,521 non-coding genes 5,363 small non-coding 14,720 long non-coding 2,222 misc non-coding Total number of transcripts can be much higher: 200,310 gene transcripts
30 RNA-seq workflow III Short read alignment Goal: determine (optimal) mapping of each sequencing read to reference SEB9BZKS1:279:C4JALACXX:8:1101:1292:2222/1 SEB9BZKS1:279:C4JALACXX:8:1101:1771:2249/1 SEB9BZKS1:279:C4JALACXX:8:1101:4645:2229/1 SEB9BZKS1:279:C4JALACXX:8:1101:4518:2229/1 SEB9BZKS1:279:C4JALACXX:8:1101:5231:2241/1 SEB9BZKS1:279:C4JALACXX:8:1101:5383:2243/1 SEB9BZKS1:279:C4JALACXX:8:1101:7221:2245/1 SEB9BZKS1:279:C4JALACXX:8:1101:8304:2249/1 SEB9BZKS1:279:C4JALACXX:8:1101:9168:2233/1 SEB9BZKS1:279:C4JALACXX:8:1101:9915:2241/1 NGTGGACAAGATTCTTGGAGCCTTACCCTTGTGTGGACCCATACCGAAGTG
 alignment")
31 RNA-seq workflow III Short read alignment Mapping = always local alignment Reads from RNA can span exons spliced (gapped) alignment necessary

32 RNA-seq workflow III Short read alignment provides three read alignment tools: RNA STAR* Advantage: one of the most sensitive, precise, versatile and fast read alignment programs Disadvantage: memory-intensive HISAT2** - fast and sensitive, can be run on a laptop TopHat*** - fast splice junction mapper, uses Bowtie2 and then analyzes the mapping results to identify splice junctions between exons genome indexes precomputed for human and mouse *Dobin et al., Bioinformatics, 2013 **Kim et al., Nature Methods, 2015 ***Kim et. al., Genome Biology, 2013
 of the six FASTQ files Select Homo_sapiens.")
33 Galaxy practical part III short read alignment Transcriptomics Mapping HISAT2 Select unpaired reads Choose one(!!!) of the six FASTQ files Select Homo_sapiens... as a reference genome Click Execute When job is scheduled click on HISAT2 again and read the description Note: mapping will take a while (~30min.)!
34 Galaxy practical part III short read alignment

35 RNA-seq workflow III Short read alignment Visualization of alignments as stacked read sequences:
36 RNA-seq workflow III Short read alignment More flexible: Genome browsers Visualization of reads, splice patterns, mutations etc. Integration of annotation, public data, known SNPs etc. UCSC online genome browser: genome.ucsc.edu Downloadable and usable from Galaxy: IGV from Broad Institute* software.broadinstitute.org/software/igv/ *Robinson et al., Nature Biotechnology, 2011

37 The RNA-seq workflow III Short read alignment
 High coverage helps to identify genomic variants Depends on sequencing")
38 RNA-seq workflow III Short read alignment Read coverage: # of reads matching a position/region Allows statements about gene expression level (RNA-seq) High coverage helps to identify genomic variants Depends on sequencing depth
39 RNA-seq workflow III Short read alignment SAM = Sequence Alignment/Map format Human-readable standard format for alignment characterization Contains general information on alignment program/parameters and reference sequence used One entry per alignment with information on location, quality and more BAM = Binary (compressed) version samtools: popular tool for SAM/BAM file manipulation


40 RNA-seq workflow III Short read alignment
41 RNA-seq workflow III Short read alignment Several metrics allow statements about the total sample alignment quality: Total number of mapped reads ( coverage) and fraction of reads mapping to the genome......uniquely: evidence for particular gene/transcript...multiply: paralogs, CNV, ribosomal RNA,......not at all: contamination, genomic DNA,... # mismatches # novel splice junctions...

42 RNA-seq workflow III Short read alignment Example mapping output: Click on the finished job and inspect the mapping statistics Click the info icon to assess information on the job details including version of the software used

43 Galaxy practical part III short read alignment Start IGV on your system (search on Desktop) Open.bat file Choose Human Hg38 as a reference genome Go to the locus field and enter PCDH7
44 Galaxy practical part III short read alignment Shared Data Data Libraries RNA-Seq_MolBio_Lecture Aligned_Files Import all alignment ( BAM ) files into your history Ignore file Aligned_PCDH7-3.bam with size Mb Go to main view ( Analyze Data ) Select one alignment file from GFP, one alignment file from PCDH7, and click display with IGV local Go to IGV, zoom in on the first exon of PCDH7 Right-click on the data tracks and choose Collapsed
45 RNAseq-workflow IV - quantification of expression Gene expression quantification Goal: estimate the gene expression level from counting reads overlapping annotated genes discoveringthegenome.org
46 RNAseq-workflow IV quantification of expression Annotations are often available from genome project websites or Ensembl Standard format for annotations is the general feature format (GFF) or gene transfer format (GTF) Tab-delimited files with information on gene structures 10 fields including flexible Attributes

47 RNAseq-workflow IV quantification of expression The file we down-/uploaded earlier is an annotation in GTF format for the human genome
 and summarize on meta-feature (here: gene)")
48 RNAseq-workflow IV - quantification of expression Standard procedure: count number of reads that overlap features (here: exons of a gene) and summarize on meta-feature (here: gene) level

49 RNAseq-workflow IV - quantification of expression Questions and pitfalls when counting mapped reads Consider multiply mapped reads? Count on gene or exon/transcript level? How to count partially mapping reads? How to treat overlapping features?...
50 RNAseq-workflow IV - quantification of expression Galaxy@GWDG provides featurecounts* tool for fast and flexible quantification Transcriptomics Counting featurecounts and read the description Click Multiple datasets and select all imported alignment files load the annotation file (the GTF file) from your history Click Execute quantification should take between 1 to 10 min. *Liao et al., Bioinformatics, 2014
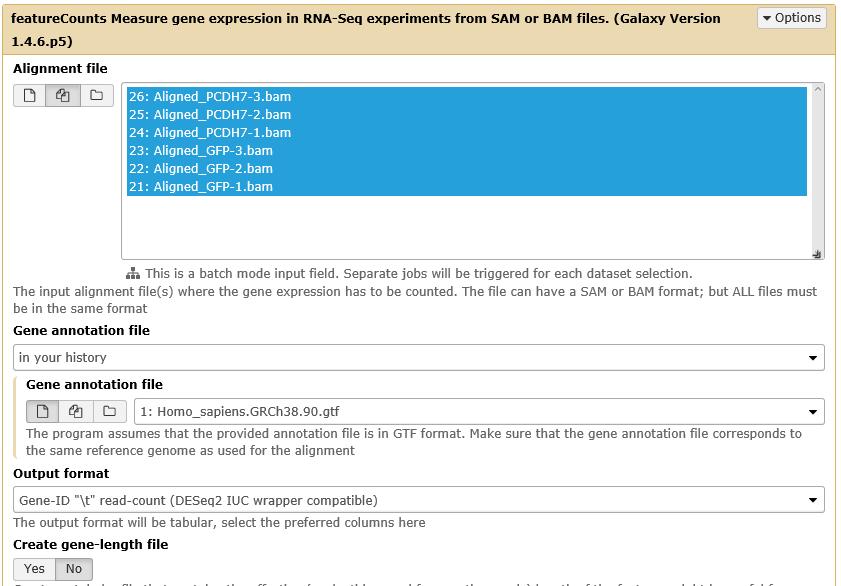
51
52 Galaxy practical part IV gene expression quantification When any dataset is finished, click on eye symbol Copy identifier of a gene with >1000 reads assigned and paste it into Ensembl search window Optional: rename files according to alignment input


53 RNA workflow addendum Summary of quality from multiple samples Quality assessment of 6 samples easy enough to do one by one What about more? Solution: MultiQC Supports summary logs from multiple software, including FastQC, STAR, Bowtie2, featurecounts, etc. Generates a single HTML file, summarizing all results in a single, interactive report

54 RNA workflow addendum Summary of quality from multiple samples

55 Galaxy practical addendum quality summary (FastQC)


56 Galaxy practical addendum quality summary Questions?
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Rsubread package: high-performance read alignment, quantification and mutation discovery
 Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Supplementary Figure 1. Fast read-mapping algorithm of BrowserGenome.
 Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
Rsubread package: high-performance read alignment, quantification and mutation discovery
 Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6
 Exercise 6") Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Genome 373: Mapping Short Sequence Reads III. Doug Fowler
 Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Analyzing Variant Call results using EuPathDB Galaxy, Part II
 Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
How to store and visualize RNA-seq data
 How to store and visualize RNA-seq data Gabriella Rustici Functional Genomics Group gabry@ebi.ac.uk EBI is an Outstation of the European Molecular Biology Laboratory. Talk summary How do we archive RNA-seq
How to store and visualize RNA-seq data Gabriella Rustici Functional Genomics Group gabry@ebi.ac.uk EBI is an Outstation of the European Molecular Biology Laboratory. Talk summary How do we archive RNA-seq
Aligners. J Fass 21 June 2017
 Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
TP RNA-seq : Differential expression analysis
 TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
Helpful Galaxy screencasts are available at:
 This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
The software comes with 2 installers: (1) SureCall installer (2) GenAligners (contains BWA, BWA- MEM).
 SureCall installer (2) GenAligners (contains BWA, BWA- MEM).") Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Resequencing Analysis. (Pseudomonas aeruginosa MAPO1 ) Sample to Insight
 Sample to Insight") Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
Services Performed. The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples.
 Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
ChIP-Seq Tutorial on Galaxy
 1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Genome Browsers - The UCSC Genome Browser
 Genome Browsers - The UCSC Genome Browser Background The UCSC Genome Browser is a well-curated site that provides users with a view of gene or sequence information in genomic context for a specific species,
Genome Browsers - The UCSC Genome Browser Background The UCSC Genome Browser is a well-curated site that provides users with a view of gene or sequence information in genomic context for a specific species,
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Creating and Using Genome Assemblies Tutorial
 Creating and Using Genome Assemblies Tutorial Release 8.1 Golden Helix, Inc. March 18, 2014 Contents 1. Create a Genome Assembly for Danio rerio 2 2. Building Annotation Sources 5 A. Creating a Reference
Creating and Using Genome Assemblies Tutorial Release 8.1 Golden Helix, Inc. March 18, 2014 Contents 1. Create a Genome Assembly for Danio rerio 2 2. Building Annotation Sources 5 A. Creating a Reference
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame
 1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Sequence Alignment. GBIO0002 Archana Bhardwaj University of Liege
 Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Integrated Genome browser (IGB) installation
 installation") Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
Advanced UCSC Browser Functions
 Advanced UCSC Browser Functions Dr. Thomas Randall tarandal@email.unc.edu bioinformatics.unc.edu UCSC Browser: genome.ucsc.edu Overview Custom Tracks adding your own datasets Utilities custom tools for
Advanced UCSC Browser Functions Dr. Thomas Randall tarandal@email.unc.edu bioinformatics.unc.edu UCSC Browser: genome.ucsc.edu Overview Custom Tracks adding your own datasets Utilities custom tools for
Long Read RNA-seq Mapper
 UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
NGS : reads quality control
 NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
The software comes with 2 installers: (1) SureCall installer (2) GenAligners (contains BWA, BWA-MEM).
 SureCall installer (2) GenAligners (contains BWA, BWA-MEM).") Release Notes Agilent SureCall 3.5 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Release Notes Agilent SureCall 3.5 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Package Rsubread. July 21, 2013
 Package Rsubread July 21, 2013 Type Package Title Rsubread: an R package for the alignment, summarization and analyses of next-generation sequencing data Version 1.10.5 Author Wei Shi and Yang Liao with
Package Rsubread July 21, 2013 Type Package Title Rsubread: an R package for the alignment, summarization and analyses of next-generation sequencing data Version 1.10.5 Author Wei Shi and Yang Liao with
Intro to NGS Tutorial
 Intro to NGS Tutorial Release 8.6.0 Golden Helix, Inc. October 31, 2016 Contents 1. Overview 2 2. Import Variants and Quality Fields 3 3. Quality Filters 10 Generate Alternate Read Ratio.........................................
Intro to NGS Tutorial Release 8.6.0 Golden Helix, Inc. October 31, 2016 Contents 1. Overview 2 2. Import Variants and Quality Fields 3 3. Quality Filters 10 Generate Alternate Read Ratio.........................................
QIAseq Targeted RNAscan Panel Analysis Plugin USER MANUAL
 QIAseq Targeted RNAscan Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted RNAscan Panel Analysis 0.5.2 beta 1 Windows, Mac OS X and Linux February 5, 2018 This software is for research
QIAseq Targeted RNAscan Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted RNAscan Panel Analysis 0.5.2 beta 1 Windows, Mac OS X and Linux February 5, 2018 This software is for research
Exercises: Analysing RNA-Seq data
 Exercises: Analysing RNA-Seq data Version 2018-03 Exercises: Analysing RNA-Seq data 2 Licence This manual is 2011-18, Simon Andrews, Laura Biggins. This manual is distributed under the creative commons
Exercises: Analysing RNA-Seq data Version 2018-03 Exercises: Analysing RNA-Seq data 2 Licence This manual is 2011-18, Simon Andrews, Laura Biggins. This manual is distributed under the creative commons
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Tutorial 4 BLAST Searching the CHO Genome
 Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
Analysis of ChIP-seq data
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Introduction to Galaxy
 Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
Tutorial. Find Very Low Frequency Variants With QIAGEN GeneRead Panels. Sample to Insight. November 21, 2017
 Find Very Low Frequency Variants With QIAGEN GeneRead Panels November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Find Very Low Frequency Variants With QIAGEN GeneRead Panels November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Using the Galaxy Local Bioinformatics Cloud at CARC
 Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University
Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL
 QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
Using Galaxy: RNA-seq
 Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Subread/Rsubread Users Guide
 Subread/Rsubread Users Guide Rsubread v1.32.3/subread v1.6.3 25 February 2019 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Subread/Rsubread Users Guide Rsubread v1.32.3/subread v1.6.3 25 February 2019 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Goal: Learn how to use various tool to extract information from RNAseq reads. 4.1 Mapping RNAseq Reads to a Genome Assembly
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Data Processing and Analysis in Systems Medicine. Milena Kraus Data Management for Digital Health Summer 2017
 Milena Kraus Digital Health Summer Agenda Real-world Use Cases Oncology Nephrology Heart Insufficiency Additional Topics Data Management & Foundations Biology Recap Data Sources Data Formats Business Processes
Milena Kraus Digital Health Summer Agenda Real-world Use Cases Oncology Nephrology Heart Insufficiency Additional Topics Data Management & Foundations Biology Recap Data Sources Data Formats Business Processes
Aligning reads: tools and theory
 Aligning reads: tools and theory Genome Sequence read :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-14pos :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg chrx: 152139280 152139290 152139300
Aligning reads: tools and theory Genome Sequence read :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-14pos :LM-Mel-42neg :LM-Mel-14neg :LM-Mel-42neg :LM-Mel-14neg chrx: 152139280 152139290 152139300
Subread/Rsubread Users Guide
 Subread/Rsubread Users Guide Subread v1.4.6-p3/rsubread v1.18.0 15 May 2015 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Subread/Rsubread Users Guide Subread v1.4.6-p3/rsubread v1.18.0 15 May 2015 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Subread/Rsubread Users Guide
 Subread/Rsubread Users Guide Rsubread v1.32.0/subread v1.6.3 19 October 2018 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Subread/Rsubread Users Guide Rsubread v1.32.0/subread v1.6.3 19 October 2018 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
AgroMarker Finder manual (1.1)
") AgroMarker Finder manual (1.1) 1. Introduction 2. Installation 3. How to run? 4. How to use? 5. Java program for calculating of restriction enzyme sites (TaqαI). 1. Introduction AgroMarker Finder (AMF)is
AgroMarker Finder manual (1.1) 1. Introduction 2. Installation 3. How to run? 4. How to use? 5. Java program for calculating of restriction enzyme sites (TaqαI). 1. Introduction AgroMarker Finder (AMF)is
Browser Exercises - I. Alignments and Comparative genomics
 Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
The software and data for the RNA-Seq exercise are already available on the USB system
 BIT815 Notes on R analysis of RNA-seq data The software and data for the RNA-Seq exercise are already available on the USB system The notes below regarding installation of R packages and other software
BIT815 Notes on R analysis of RNA-seq data The software and data for the RNA-Seq exercise are already available on the USB system The notes below regarding installation of R packages and other software
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq
 TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
Genome Browsers Guide
 Genome Browsers Guide Take a Class This guide supports the Galter Library class called Genome Browsers. See our Classes schedule for the next available offering. If this class is not on our upcoming schedule,
Genome Browsers Guide Take a Class This guide supports the Galter Library class called Genome Browsers. See our Classes schedule for the next available offering. If this class is not on our upcoming schedule,
Tutorial: RNA-Seq Analysis Part II (Tracks): Non-Specific Matches, Mapping Modes and Expression measures
: Non-Specific Matches, Mapping Modes and Expression measures") : RNA-Seq Analysis Part II (Tracks): Non-Specific Matches, Mapping Modes and February 24, 2014 Sample to Insight : RNA-Seq Analysis Part II (Tracks): Non-Specific Matches, Mapping Modes and : RNA-Seq Analysis
: RNA-Seq Analysis Part II (Tracks): Non-Specific Matches, Mapping Modes and February 24, 2014 Sample to Insight : RNA-Seq Analysis Part II (Tracks): Non-Specific Matches, Mapping Modes and : RNA-Seq Analysis
Understanding and Pre-processing Raw Illumina Data
 Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Accessible, Transparent and Reproducible Analysis with Galaxy
 Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
de.nbi and its Galaxy interface for RNA-Seq
 de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
Exercise 2: Browser-Based Annotation and RNA-Seq Data
 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Fusion Detection Using QIAseq RNAscan Panels
 Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Reference guided RNA-seq data analysis using BioHPC Lab computers
 Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further