RNA-seq. Manpreet S. Katari
|
|
|
- Bertram Preston
- 6 years ago
- Views:
Transcription
1 RNA-seq Manpreet S. Katari

2 Evolution of Sequence Technology
3
4 Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene (sum of exons / 1000) T = total number of reads in the library mapped to the genome / 1,000,000
5 Illumina mrna sequencing Log (KNO3/KCl) N-regulation of mrna: Illumina vs ATH1 R 2 = 0.85 Log (KNO3/KCl) Affymetrix ATH1 chips


6 Detection of Arabidopsis Genes pre-mirna Annotated cdnas 4,892 detected by Illumina 22,173 14, , Affymetrix Unambiguously detected genes: 14,725 Genes on Affy chips with Absent Call
7 RNA-seq provides even more
8 RNA-seq pipeline Manpreet S. Katari
9 The basic workflow 1. Evaluate the quality of the sequences a. Use fastqc to asses quality of sequence 2. Trim low quality sequences a. Use fastx tool kit 3. Map the reads to the Genome a. Build the bowtie2 database b. Run the alignment using tophat2 4. Link mapped reads into genes and calculate normalized expression values a. Use cufflinks to determine normalized values of each run. 5. Compare samples to determine differentially expressed genes. a. Use cuffdiff to compare the different samples and identify differentially expressed genes.
10 Processing RNA-seq reads (Filter) Remove not so interesting RNA molecules Majority of the RNA molecules in the cell are ribosomal rna. Low complexity sequences For example PolyA sequences. Adapter sequences Occasionally some of the reads can contain adapter sequences. Illumina reads have tendency to have poor quality reads in the 3 Trim reads on either end and also based on quality.

11 Read Identifier Fastq format Read Sequence Read Sequence Quality
12 Module environment review To look at the different modules available: module avail To load a module module load fastqc To get a list of modules already loaded module list To remove or unload a module module unload fastqc To get help on fastqc fastqc -h
13 1. Perform Quality control We will use the Fastqc package to evaluate the quality of our sequences. module load fastqc/ fastqc sequence.fastq Transfer the folder to your local machine to view results. On the pc we used the sftp on mobaxterm Another option is WinSCP ( On the mac you can use cyberduck or scp commad Extract the folder and open the fastqc_report.html
14 Cleaning up Once you have the files on your computer and you don t need it on the server simply delete them. The r option allows you to delete recursively all files and directories below the one provided.
15 2. Trimming the reads - Sequences generated from illumina platform tend to have lower quality sequences specially at the 3' end. - Since our sequence alignment algorithms are looking for nearly exact match, we want to trim the sequence from 5 and 3 end. - Our sequence is 50bp, so let's trim 5 from 5 and stop at base We also noticed that - We will use a software available for free called fastx o module load fastx_toolkit/ fastx_trimmer f 5 sequence.fastq -l 40 o sequence.trimmed.fastq fastx_clipper -a ATCGTATGCCGTCTTCTGCTTG -l 25 I sequence.trimmed.fastq o sequence_trimmed_clipped.fastq
16 Aligning Short reads

17 New Algorithms for short sequences
18 Two main types of alignment methods Hash-table based Burrows and Wheeler Transformation Both can be applied to Illumina and Solid Both start with different heuristics to reduce the search space but then finally use a more accurate alignment method like Smith Waterman. 18
19 Hash Table (BLAT) 19
20 Burrows Wheeler Transformation Li and Durbin (2009) Bioinformatics 20
21 Which is better? BWA is about 10x faster then hash-based methods and takes less memory. BWA is less sensitive. Based on the query size it can only allow a given number of mismatches For example for 100bp max of 5 mismatch. 21
22 Mapping Reads from RNA molecules What is the advantage of mapping reads from RNA to the genome sequenced instead of a database of all predicted RNA molecules? We are not depending on the quality of annotation. We are not assuming that we know about all of the RNA molecules in the cell. How can we find reads mapping to spliced junctions? Create a separate database of all possible spliced junctions Split reads in half and map them separately.
23 Bowtie & TopHat Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012, 9:
24 3. Mapping the reads To align our sequences to the genome we will use the Bowtie-Tophat algorithm discussed in class 1. Building the database a. In order to use bowtie and tophat for our analysis we have to first create the database. b. The following command will create a database in your current directory module load bowtie2/2.2.2 bowtie2-build /home/mkatari/arabidopsis.fa Arabidopsis
25 3. Mapping the reads 2. Run the alignment module load tophat2/ tophat2 -i 20 -I o tophat_output \ /home/mkatari/nitrogen/arabidopsis \ sequence_trimmed_clipped.fastq -i = minimum intron size -I = maximum intron size -o = output directory Database Query file

26 Tophat result: sam file

27 SAM (Sequence Alignment Map) Popular output format pysam - An interface for reading and writing SAM files

28 Bowtie output (SAM) 1. HYYD8:00007: gb CM M1D9M1D9M2D21M2D18M1D70M 7. * CAATGAGCTAACAACTGCAATGGGGCCATAATGGCTGCTTGTCGTTTGGCACGTACATGGACTAGCTTCC CCCGTGGCACAAAAATGGCTCTACGTTCTGTTACGAGCGCACCTACTGAAGGTCTCTCATAGGAGTGTAT GTATATGCATATACAT 11. ;:=>>:333*33,33<<:7:3*344, >>::4-6666B<EB>ABA@?;::44,4444<<4,4* ??670??==?<?@?>>>><7<<45-??>>?>>>??;< ,:;;< ?667?=@@888@AA@?<>;< AS:i:-58 XN:i:0 XM:i:4 XO:i:5 XG:i:7 NM:i:11 MD:Z:29^A9^T9^TG10C0T1G0A6^CC18^A70 YT:Z:UU XR:Z:@HYYD8%3A00007%3A00087%0AATGTATATGCATATACATACACTCCTATGAGAGACCTTCAGT AGGTGCGCTCGTAACAGAACGTAGAGCCATTTTTGTGCCACGGGGGAAGCTAGTCCATGTACGTGCCAA ACGACAAGCAGCCATTATGGCCCCATTGCAGTTGTTAGCTCATTG%0A+%0A55<;><?@AA@888@@ =?766? <;;%3A, <;??>>>?>>??-54<<7<>>>>?@?<?==??076?? *4,4<<4444,44%3A%3A;?@ABA>BE<B6666-4%3A%3A>> ,443*3%3A7%3A<<33,33*333%3A>>=%3A;%0A

29 Bitwise Flag What is 77? Find greatest value without going over = = = = 0 1 What is 141?

30 CIGAR string 29M 1D 9M 1D 9M 2D 21M 2D 18M 1D 70M

31 Cufflinks first starts with the output of any alignment tool such as TopHat

32 Then it assembles the isoforms by first identifying the reads that can not be assembled together.

33 Then calculate abundance

34 Assembling the reads to identify transcripts.
35 CuffCompare The program cuffcompare helps you: Compare your assembled transcripts to a reference annotation Track Cufflinks transcripts across multiple experiments (e.g. across a time course) Output contains codes = match c contained j new isoform u unknown, intergenic transcript i single exon in intron region
36 Identification of spliced junctions depends largely on the depth of sequences coverage.
37 4. Link genes and determine normalized expression values Run cufflinks. We will enter the tophat output directory and run it in there so all cufflinks output will be in tophat_output module load cufflinks/2.2.1 cd tophat_output cufflinks -G /home/mkatari/manny/arabidosis.gtf \ accepted_hits.sam -o = output directory -G = GTF file sam output file

38 GTF files are like GFF file but with specific attributes If you find a gff file for your organism, you can easily convert it into a gtf file. TAIR10_GFF3_genes.gff is a file I downloaded from TAIR which contains coordinates of the genes annotated in Arabidopsis. gffread -E TAIR10_GFF3_genes.gff -T -o- > Arabidopsis.gtf

39 Cufflinks output: fpkm normalized values
40 Cuffdiff Can be use to find significant changes in transcript expression, splicing, and promoter use. Inputs are: Annotation to compare (can be output from cufflinks) Tophat output from different samples Cuffdiff allows to compare samples even if you have only replicate.
41 5. Compare expression values of two sample Run cuffdiff - data is normalized and a modified version of t-test is used and p-values are corrected for multiple hypothesis testing. cuffdiff o cuff_diff \ L KCL,NO3 \ -dispersion-method poisson \ -library-norm-method quartile \ /home/mkatari/nitrogen/arabidopsis.gtf \ /home/mkatari/nitrogen/kcl1/accepted_hits.bam,/home/mkatari/nitrogen/kcl2/accepted_hits.bam \ /home/mkatari/nitrogen/no31/accepted_hits.sam,/home/mkatari/nitrogen/no32/accepted_hits.bam


42 Cuffdiff results To get a list of genes that are significantly differentially expressed genes cut -f 1,8,9,10,12,13,14 gene_exp.diff grep "yes" less
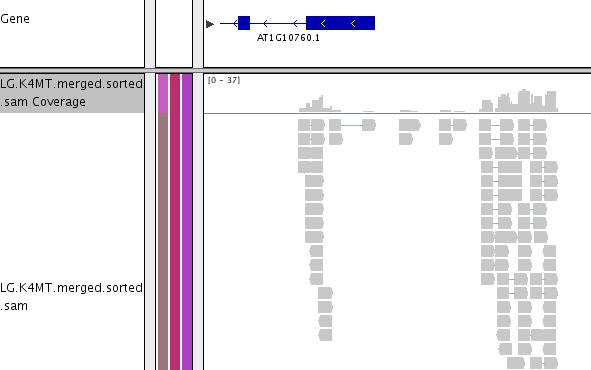
43 IGV
44 IGV: Integrative Genomics Viewer Standalone java program Does not require a mysql database server or an apache web server Limited to the resources of the machine that it is running on. More interactive compared to Gbrowse. Both IGV and Gbrowse can use GFF file format.
45 IGV tools Simple tools to format the files so you can use them on the browser Tools that I have needed: Sort Index
46 Visualizing in IGV There are two main steps: You have to index the reference sequence Sort and index the bam files module load samtools/ samtools faidx Arabidopsis.fa samtools sort KCL1/accepted_hits.bam KCL1_sorted.bam samtools index KCL1_sorted.bam samtools sort KCL2/accepted_hits.bam KCL2_sorted.bam samtools index KCL1_sorted.bam samtools sort NO31/accepted_hits.sam NO31_sorted.bam samtools index KCL1_sorted.bam samtools sort NO32/accepted_hits.bam NO32_sorted.bam samtools index KCL1_sorted.bam
47 Launch IGV module load module load igv/ igv First load the genome fasta sequence File->Load Genome from File Select the Arabidopsis.fa in /home/mkatari/nitrogen/ Then load the annotations File->Load from File Select the Arabidopsis.gft in /home/mkatari/nitrogen/ Then load as many alignments you would like File->Load from File Select the KCL1_sorted.bam in /home/mkatari/nitrogen/ IGV is also capable of visualizing VCF(Variant call format)
48 Don t forget to Dedup!! Sequencing platforms are not perfect. Occasionally one dna fragment can generate many clusters. You know you have this problem when the reads are exactly the same and are generated using random sequencing. module load samtools/ samtools sort KCL1/accepted_hits.bam KCL1/accepted_hits_sorted samtools rmdup -s KCL1/accepted_hits_sorted.bam KCL1/accepted_dedup.bam
49 Assignment Repeat the Cuffdiff step using dedup alignment files How many genes are differentially expressed now? Repeat the sorting and indexing for the deduped files so we can visualize them on IGV. Make sure the artifacts have all gone. Confirm results by looking at AT1G77760 Is it higher or lower in the presence of Nitrate?
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Introduction to Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
TP RNA-seq : Differential expression analysis
 TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Aligners. J Fass 21 June 2017
 Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
version /1/2011 Source code Linux x86_64 binary Mac OS X x86_64 binary
 Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv
 Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat a.tgz. Software:
 A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Services Performed. The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples.
 Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Goal: Learn how to use various tool to extract information from RNAseq reads. 4.1 Mapping RNAseq Reads to a Genome Assembly
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
TopHat, Cufflinks, Cuffdiff
 TopHat, Cufflinks, Cuffdiff Andreas Gisel Institute for Biomedical Technologies - CNR, Bari TopHat TopHat TopHat TopHat is a program that aligns RNA-Seq reads to a genome in order to identify exon-exon
TopHat, Cufflinks, Cuffdiff Andreas Gisel Institute for Biomedical Technologies - CNR, Bari TopHat TopHat TopHat TopHat is a program that aligns RNA-Seq reads to a genome in order to identify exon-exon
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
RNASeq2017 Course Salerno, September 27-29, 2017
 RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Gene expression estimation
 Gene expression estimation") mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
Aligners. J Fass 23 August 2017
 Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Reference guided RNA-seq data analysis using BioHPC Lab computers
 Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Evaluate NimbleGen SeqCap RNA Target Enrichment Data
 Roche Sequencing Technical Note November 2014 How To Evaluate NimbleGen SeqCap RNA Target Enrichment Data 1. OVERVIEW Analysis of NimbleGen SeqCap RNA target enrichment data generated using an Illumina
Roche Sequencing Technical Note November 2014 How To Evaluate NimbleGen SeqCap RNA Target Enrichment Data 1. OVERVIEW Analysis of NimbleGen SeqCap RNA target enrichment data generated using an Illumina
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
File Formats: SAM, BAM, and CRAM. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
Long Read RNA-seq Mapper
 UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
Our data for today is a small subset of Saimaa ringed seal RNA sequencing data (RNA_seq_reads.fasta). Let s first see how many reads are there:
. Let s first see how many reads are there:") Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
Tiling Assembly for Annotation-independent Novel Gene Discovery
 Tiling Assembly for Annotation-independent Novel Gene Discovery By Jennifer Lopez and Kenneth Watanabe Last edited on September 7, 2015 by Kenneth Watanabe The following procedure explains how to run the
Tiling Assembly for Annotation-independent Novel Gene Discovery By Jennifer Lopez and Kenneth Watanabe Last edited on September 7, 2015 by Kenneth Watanabe The following procedure explains how to run the
Read Mapping. Slides by Carl Kingsford
 Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
KisSplice. Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data. 29th may 2013
 Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)
Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)
11/8/2017 Trinity De novo Transcriptome Assembly Workshop trinityrnaseq/rnaseq_trinity_tuxedo_workshop Wiki GitHub
 trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
Exercise 1 Review. --outfiltermismatchnmax : max number of mismatch (Default 10) --outreadsunmapped fastx: output unmapped reads
 --outreadsunmapped fastx: output unmapped reads") Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide
 RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq
 TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
Variation among genomes
 Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Using Galaxy: RNA-seq
 Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
RNAseq analysis: SNP calling. BTI bioinformatics course, spring 2013
 RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
Questions about Cufflinks should be sent to Please do not technical questions to Cufflinks contributors directly.
 Cufflinks RNA-Seq analysis tools - User's Manual 1 of 22 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Please Note If you have questions
Cufflinks RNA-Seq analysis tools - User's Manual 1 of 22 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Please Note If you have questions
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
A Virtual Machine to teach NGS data analysis. Andreas Gisel CNR - ITB Bari, Italy
 A Virtual Machine to teach NGS data analysis Andreas Gisel CNR - ITB Bari, Italy The Virtual Machine A virtual machine is a tightly isolated software container that can run its own operating systems and
A Virtual Machine to teach NGS data analysis Andreas Gisel CNR - ITB Bari, Italy The Virtual Machine A virtual machine is a tightly isolated software container that can run its own operating systems and
replace my_user_id in the commands with your actual user ID
 Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
WM2 Bioinformatics. ExomeSeq data analysis part 1. Dietmar Rieder
 WM2 Bioinformatics ExomeSeq data analysis part 1 Dietmar Rieder RAW data Use putty to logon to cluster.i med.ac.at In your home directory make directory to store raw data $ mkdir 00_RAW Copy raw fastq
WM2 Bioinformatics ExomeSeq data analysis part 1 Dietmar Rieder RAW data Use putty to logon to cluster.i med.ac.at In your home directory make directory to store raw data $ mkdir 00_RAW Copy raw fastq
Analysis of ChIP-seq data
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
RNA- SeQC Documentation
 RNA- SeQC Documentation Description: Author: Calculates metrics on aligned RNA-seq data. David S. DeLuca (Broad Institute), gp-help@broadinstitute.org Summary This module calculates standard RNA-seq related
RNA- SeQC Documentation Description: Author: Calculates metrics on aligned RNA-seq data. David S. DeLuca (Broad Institute), gp-help@broadinstitute.org Summary This module calculates standard RNA-seq related
Connect to login8.stampede.tacc.utexas.edu. Sample Datasets
 Alignment Overview Connect to login8.stampede.tacc.utexas.edu Sample Datasets Reference Genomes Exercise #1: BWA global alignment Yeast ChIP-seq Overview ChIP-seq alignment workflow with BWA Introducing
Alignment Overview Connect to login8.stampede.tacc.utexas.edu Sample Datasets Reference Genomes Exercise #1: BWA global alignment Yeast ChIP-seq Overview ChIP-seq alignment workflow with BWA Introducing
Short Read Sequencing Analysis Workshop
 Short Read Sequencing Analysis Workshop Day 8: Introduc/on to RNA-seq Analysis In-class slides Day 7 Homework 1.) 14 GABPA ChIP-seq peaks 2.) Error: Dataset too large (> 100000). Rerun with larger maxsize
Short Read Sequencing Analysis Workshop Day 8: Introduc/on to RNA-seq Analysis In-class slides Day 7 Homework 1.) 14 GABPA ChIP-seq peaks 2.) Error: Dataset too large (> 100000). Rerun with larger maxsize
Identiyfing splice junctions from RNA-Seq data
 Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
From the Schnable Lab:
 From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
RNA Sequencing with TopHat and Cufflinks
 RNA Sequencing with TopHat and Cufflinks Introduction 3 Run TopHat App 4 TopHat App Output 5 Run Cufflinks 18 Cufflinks App Output 20 RNAseq Methods 27 Technical Assistance ILLUMINA PROPRIETARY 15050962
RNA Sequencing with TopHat and Cufflinks Introduction 3 Run TopHat App 4 TopHat App Output 5 Run Cufflinks 18 Cufflinks App Output 20 RNAseq Methods 27 Technical Assistance ILLUMINA PROPRIETARY 15050962
SAM : Sequence Alignment/Map format. A TAB-delimited text format storing the alignment information. A header section is optional.
 Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
New releases and related tools will be announced through the mailing list
 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Please Note If you have questions about how to use Cufflinks or would like more information about the software,
Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Please Note If you have questions about how to use Cufflinks or would like more information about the software,
RCAC. Job files Example: Running seqyclean (a module)
") RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Read Mapping and Assembly
 Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Scalable RNA Sequencing on Clusters of Multicore Processors
 JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Visualization using CummeRbund 2014 Overview
 Visualization using CummeRbund 2014 Overview In this lab, we'll look at how to use cummerbund to visualize our gene expression results from cuffdiff. CummeRbund is part of the tuxedo pipeline and it is
Visualization using CummeRbund 2014 Overview In this lab, we'll look at how to use cummerbund to visualize our gene expression results from cuffdiff. CummeRbund is part of the tuxedo pipeline and it is
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers
 Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Fast-track to Gene Annotation and Genome Analysis
 Fast-track to Gene Annotation and Genome Analysis Contents Section Page 1.1 Introduction DNA Subway is a bioinformatics workspace that wraps high-level analysis tools in an intuitive and appealing interface.
Fast-track to Gene Annotation and Genome Analysis Contents Section Page 1.1 Introduction DNA Subway is a bioinformatics workspace that wraps high-level analysis tools in an intuitive and appealing interface.
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Ballgown. flexible RNA-seq differential expression analysis. Alyssa Frazee Johns Hopkins
 Ballgown flexible RNA-seq differential expression analysis Alyssa Frazee Johns Hopkins Biostatistics @acfrazee RNA-seq data Reads (50-100 bases) Transcripts (RNA) Genome (DNA) [use tool of your choice]
Ballgown flexible RNA-seq differential expression analysis Alyssa Frazee Johns Hopkins Biostatistics @acfrazee RNA-seq data Reads (50-100 bases) Transcripts (RNA) Genome (DNA) [use tool of your choice]
Benchmarking of RNA-seq aligners
 Lecture 17 RNA-seq Alignment STAR Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Based on this analysis the most reliable
Lecture 17 RNA-seq Alignment STAR Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Based on this analysis the most reliable
preparation methods and new bacterial strains. Parts of the pipeline that can be updated will be annotated in this guide.
 BacSeq Introduction The purpose of this guide is to aid current and future Whiteley Lab members and University of Texas microbiologists with bacterial RNA?Seq analysis. Once you have analyzed your data
BacSeq Introduction The purpose of this guide is to aid current and future Whiteley Lab members and University of Texas microbiologists with bacterial RNA?Seq analysis. Once you have analyzed your data
Welcome to GenomeView 101!
 Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Genome-wide analysis of degradome data using PAREsnip2
 Genome-wide analysis of degradome data using PAREsnip2 07/06/2018 User Guide A tool for high-throughput prediction of small RNA targets from degradome sequencing data using configurable targeting rules
Genome-wide analysis of degradome data using PAREsnip2 07/06/2018 User Guide A tool for high-throughput prediction of small RNA targets from degradome sequencing data using configurable targeting rules
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching
 SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
Genome-wide analysis of degradome data using PAREsnip2
 Genome-wide analysis of degradome data using PAREsnip2 24/01/2018 User Guide A tool for high-throughput prediction of small RNA targets from degradome sequencing data using configurable targeting rules
Genome-wide analysis of degradome data using PAREsnip2 24/01/2018 User Guide A tool for high-throughput prediction of small RNA targets from degradome sequencing data using configurable targeting rules
Today's outline. Resources. Genome browser components. Genome browsers: Discovering biology through genomics. Genome browser tutorial materials
 Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection
 CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
Circ-Seq User Guide. A comprehensive bioinformatics workflow for circular RNA detection from transcriptome sequencing data
 Circ-Seq User Guide A comprehensive bioinformatics workflow for circular RNA detection from transcriptome sequencing data 02/03/2016 Table of Contents Introduction... 2 Local Installation to your system...
Circ-Seq User Guide A comprehensive bioinformatics workflow for circular RNA detection from transcriptome sequencing data 02/03/2016 Table of Contents Introduction... 2 Local Installation to your system...
Centre (CNIO). 3rd Melchor Fernández Almagro St , Madrid, Spain. s/n, Universidad de Vigo, Ourense, Spain.
. 3rd Melchor Fernández Almagro St , Madrid, Spain. s/n, Universidad de Vigo, Ourense, Spain.") O. Graña *a,b, M. Rubio-Camarillo a, F. Fdez-Riverola b, D.G. Pisano a and D. Glez-Peña b a Bioinformatics Unit, Structural Biology and BioComputing Programme, Spanish National Cancer Research Centre (CNIO).
O. Graña *a,b, M. Rubio-Camarillo a, F. Fdez-Riverola b, D.G. Pisano a and D. Glez-Peña b a Bioinformatics Unit, Structural Biology and BioComputing Programme, Spanish National Cancer Research Centre (CNIO).
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units
 GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
Integrated Genome browser (IGB) installation
 installation") Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
Integrated Genome browser (IGB) installation Navigate to the IGB download page http://bioviz.org/igb/download.html You will see three icons for download: The three icons correspond to different memory
all M 2M_gt_15 2M_8_15 2M_1_7 gt_2m TopHat2
 Pairs processed per second 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 72,318 418 1,666 49,495 21,123 69,984 35,694 1,9 71,538 3,5 17,381 61,223 69,39 55 19,579 44,79 65,126 96 5,115 33,6 61,787
Pairs processed per second 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 6, 4, 2, 72,318 418 1,666 49,495 21,123 69,984 35,694 1,9 71,538 3,5 17,381 61,223 69,39 55 19,579 44,79 65,126 96 5,115 33,6 61,787
Gene Expression Data Analysis. Qin Ma, Ph.D. December 10, 2017
 1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior