klinischen Datenanforderungen in Europa: Ist die Literaturroute noch möglich?
|
|
|
- Moris Phelps
- 6 years ago
- Views:
Transcription

1 Regulatorische Änderungen bei den klinischen Datenanforderungen in Europa: Ist die Literaturroute noch möglich? Hamburg, Dr. Bassil Akra Director Clinical Centre of Excellence TÜV SÜD Product Service





2 Content Current Clinical Data Requirements Clinical Evaluation - MEDDEV Examples Pre- & Post-Market Clinical Investigation New Medical Device Regulation TÜV SÜD Product Service GmbH Slide 2
3 Requirements for Clinical Data Dir. 2007/47/EC MEDDEV Rev. 3: Clinical i l evaluation MEDDEV 2.7/4: Guidelines on Clinical Investigation EN ISO 14155:2011: Clinical investigations on medical devices for human subjects MEDDEV Rev. 2: Guidelines on Post Market Clinical Follow-Up Studies TÜV SÜD Product Service Slide 3
4 Current Requirements for Clinical Data Dir. 2007/47/EC Which h medical devices require a clinical i l evaluation? ALL Medical Devices regardless of Classification Annex I: Demonstration of conformity with the essential requirements must include a clinical evaluation in accordance with Annex X TÜV SÜD Product Service Slide 4
5 Clinical Evaluation according to MEDDEV Rev. 3 Clinical Evaluation The assessment and analysis of clinical data pertaining to a medical device to verify the clinical safety and performance of the device when used as intended by the manufacturer. TÜV SÜD Product Service Slide 5



6 Clinical Evaluation according to current MEDDEV Rev. 3 Which route should you follow for the device under consideration? Equivalence Route Clinical Investigation Route TÜV SÜD Product Service Slide 6





7 Current Clinical Investigation Route Requirements MEDDEV 2.7/4 When should a clinical investigation be undertaken? The Conformity Assessment process for active implantable medical devices as well as for class III and implantable medical devices requires that a clinical investigation is undertaken unless it is duly justified to rely on existing data. Section 1.2 of Annex 7 of directive 90/385/EEC Section I.1a of Annex X of directive 93/42/EEC Risk Innovation TÜV SÜD Product Service / Slide 7



8 Current Clinical Investigation Route Requirements MEDDEV 2.7/4 When should a clinical investigation be undertaken? Depending on clinical claims, risk management outcome and on the results of the clinical evaluation, clinical investigations may also have to be performed for non- implantable medical devices of classes I, IIa and IIb. Additional clinical investigations may be feasible to corroborate the existing clinical evidence with regard to aspects of clinical performance, safety, benefit/risk-ratioratio or to determine relative effectiveness and safety with suitable comparators. TÜV SÜD Product Service / Slide 8
9 State of the Art A documented and systematic literature review A detailed discussion covering the benefit/risk ratio of the device when comparing to: All current available therapeutic alternatives for the same indication Conventional Therapy, Mdi Medical Devices and Mdii Medicinal Products Similar products from per example competitors for the same indication TÜV SÜD Product Service Slide 9





10 Clinical Evaluation RISK MANAGEMENT PROCESS Which route should you follow for the device under consideration? IDEA RISK IDENTIFICATION METHODS RISK MITIGATION METHODS RESIDUAL RISKS ACCEPTABILITY TÜV SÜD Product Service Slide 10
11 Clinical Evaluation according to MEDDEV Rev. 3 Determination of Scope Identification of Clinical Data Appraisal Analysis of relevant Data Clinical Evaluation Report TÜV SÜD Product Service Slide 11


12 Equivalence Approach based on current available literature Same intended use (Clinical condition/disease, Severity, Application Site, Patient Population, Critical Performance Parameter) + Technical and biological equivalence + No clinically significant difference regarding safety and performance TÜV SÜD Product Service Slide 12
13 Clinical Evaluation Example 1 Which route should you follow for the device under consideration? New Device Example of available competitors TÜV SÜD Product Service Slide 13



14 Clinical Evaluation Example 2 Which route should you follow for the device under consideration? A New Device B Example of available competitors TÜV SÜD Product Service Slide 14
15 Clinical Evaluation Example 3 Which route should you follow for the device under consideration? Equipment under Control Motor Patient TÜV SÜD Product Service Slide 15


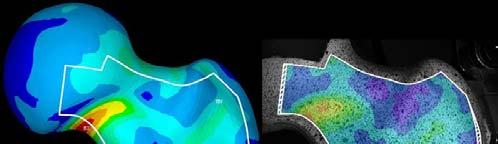

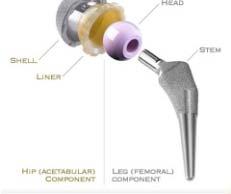
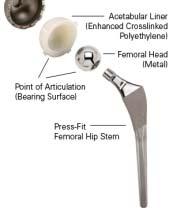
16 Equivalence Approach based on current available literature th t i / h/ t /d i hi l t Criteria Example 1 Example 2 Same intended use YES YES but possibly different population Technical Equivalence YES YES (B) NO (A) A is a short stem with significant different biomech. properties Biological Equivalence YES YES (B) NO (A) Difference Regarding Safety NO NO (A) YES (B) Difference Regarding Performance NO NO (B) YES (A) TÜV SÜD Product Service Slide 16

 NO (A)")
 YES")
 YES (A) YES NO TÜV SÜD Product")
17 Equivalence Approach based on current available literature Criteria Example 1 Example 2 Same intended use YES YES Technical Equivalence YES YES (B) NO (A) Biological Equivalence YES YES (B) NO (A) Difference Regarding Safety NO NO (A) YES (B) Difference Regarding Performance NO NO (B) YES (A) YES NO TÜV SÜD Product Service Slide 17
18 Clinical Evaluation Example 3 It is normally based on a combination of critical evaluation of the relevant scientific literature and testing according to IEC Equipment under Control Motor Patient analyze set safe state Sensor Control Actor TÜV SÜD Product Service Safety System Slide 18
19 Clinical Evaluation Example 3 Infusion pumps The main fnctionof function of the device is to pmpfl pump fluid. Functional safety considers potential hazards related to this function: Wrong flow rate Wrong volume infused Unintended start or stop of infusion Buildup of excessive pressure Air infusion Reverse flow direction To develop the device in such a way, that it can not cause any harm in case of a breakdown TÜV SÜD Product Service Slide 19
20 Good Clinical Practice Scope The ISO 14155:2011 addressess good clinical practice for the design, conduct, recording and reporting of clinical investigations carried out in human subjects to assess the safety or performance of medical devices for regulatory purposes. TÜV SÜD Product Service Slide 20


21 Good Clinical Practice How to plan a clinical investigation correctly? Risk Analysis Endpoints Scientific validity All relevant legal and regulatory requirements Clinical evaluation Study Plan Suitability Clinical relevance TÜV SÜD Product Service Slide 21
22 Clinical Investigation Plan TÜV SÜD Product Service Slide 22
23 Clinical Investigation Route Study design Type Measures to avoid bias Primary and secondary endpoints with rationale for selection and measurement Methods and timing for assessing, recording, and analyzing variables Justification for choice of comparator(s) Subjects (in-, exclusion criteria, number, duration) Study related procedures incl. post-study medical care Monitoring plan TÜV SÜD Product Service Slide 23







24 Clinical Investigation Route Statistical Methods design design, method and analytical procedures sample size level of significance expected dropout rates pass/fail criteria criteria for termination on statistical g grounds provisions for an interim analysis What is most important statistical or clinical significance? TÜV SÜD Product Service Slide 24
25 Approval in the European Union The first involved NCA provides a EUDAMED No. This number must be used for all further submissions in EU, some NCA provide an additional registration number Pre -Market NCA & EC application / approval But in some countries - exceptions for low risk studies Post-Market Out of scope => NCA & EC application / approval Within scope => EC application / approval But in some countries - if additional burden => NCA & EC application / approval TÜV SÜD Product Service Slide 25
26 Clinical Investigation Route Documents evaluated by EU Notified Bodies CIP Ltt Letter of No Objection Approval of Competent tath Authority Ethics Committee opinion Signed and dated final report TÜV SÜD Product Service Slide 26
27 PMCF Requirements according to current MEDDEV 2.12/2 Rev. 2 Why PMCF? Rare complications or problems become apparent after wide-spread or long term use of the device An appropriate PMS Plan is key to identifying and investigating residual risks associated with the use of medical devices placed on the market To confirm the safety and performance throughout the expected lifetime of the device, the continued acceptability of identified risks and to detect emerging risks on the basis of factual evidence TÜV SÜD Product Service Slide 27
28 MEDDEV 2.12/2 Rev. 2 Why PMCF? There may be limitations to the clinical data available in the pre-market phase: Duration Number of subjects Investigators involved (Heterogenicity?) Sbj Subjects (Ht (Heterogenicity?) iit?) The controlled setting of a clinical investigation vs. the full range of clinical conditions encountered in general medical practice TÜV SÜD Product Service Slide 28
29 PMCF required PMCF is mandatory for: Innovative Products Significant changes High product related risks High risk anatomical locations/target populations Emergence of new information on safety or performance Where CE marking was based on equivalence Unanswered questions of long-term safety and performance TÜV SÜD Product Service Slide 29
30 Responsibilities of a EU Notified Body The Notified Body shall Review the appropriateness of the manufacturer s general PMS procedures and plans Verify that PMCF is conducted by or on behalf of the manufacturer by appropriately competent assessors Verify that Clinical Investigations were conducted in accordance with relevant provisions of the directive(s), related guidance and standards Verify the need for PMCF and/or appropriateness of any justification Assess the appropriateness of the proposed PMCF plan TÜV SÜD Product Service Slide 30
31 Responsibilities of a EU Notified Body The Notified Body shall Verify if the gathered PMCF Data were used to update the clinical evaluation report Consider whether based on the specific device assessment, data obtained from PMCF should be transmitted between scheduled assessment activities Consider an appropriate period for certification of the product in order to set a particular time point at which PMCF data will be assessed Set conditions to submit interim reports between certification reviews TÜV SÜD Product Service Slide 31
32 Clinical Evaluation Report (CER) The appropriate CER shall be prepared as a stand-alone document to facilitate the assessment of a third independent party The stand-alone document must comprise: Product description Background regarding device technology Intended Use, Indications and Contraindications Claims regarding performance and safety Relevant bench- and preclinical data Nature and extent of clinical data Suitability of the referenced information Acceptability of the risk/benefit ratio PMCF Plan The appropriate CER must be: Signed and dated by the evaluator(s) The appropriate CER must include: A justification for the choice of evaluator(s) TÜV SÜD Product Service Slide 32


33 Where are we heading to? Rec. 2013/ /2013 MEDDEV Rev /47EG MDD AIMD IVDD To establish a robust, transparent, t predictable and sustainable regulatory framework for medical devices which ensures a high level of safety and health whilst supporting Innovation TÜV SÜD Product Service Slide 33
34 EU Regulations Proposal Scrutiny Procedure A new expert group to: Proposal Commission Medical Device Coordination Group - Made up of members appointed by the Member States and chaired by the Commission - Achieve harmonized implementation of this regulation Proposed Scrutiny Procedure (Article 44) - Establish an additional scrutiny mechanism Medical Devices Directives revision proposal Consider Manufacturer Notified Notified Body Notified Body Pre- Notified Body Submission Body Review report Approval Board CE Commission Scrutiny? Scrutiny Start Medical Devices Coordination Group MDCG T= 28 Days MDCG final comment MDCG Scrutiny T= 60 Days TÜV SÜD Product Service Slide 34
35 EU Regulations Proposal Scrutiny Procedure Proposal Parliament Manufacturer Submission SNB SNB Evaluation SNB Approval Board CE Follow Commission Scrutiny? MDCG T= 20 Days If scrutiny Opinion Appeal procedure Evaluation of CER, PMCF and other relevant doc T= 30 Days* *Clock Stop System TÜV SÜD Product Service Slide 35


36 EU Regulations Proposal Scrutiny Procedure Council of the European Union Manufacturer Submission NB Notification to the authorities Art 42(2a) EUDAMED Automatically Expert Panel* 60 Days 1. Summary of safety and performance 2. NB Assessment Report 3. Instructions ti for Use 4. Expert Panel Decision 5. Justification of NB in case of divergent views Competent authority or Commission may apply appropriate measures Monitoring and assessment of NB Review NB Assessment Changes to designations and notifications Challenge to the competence of NB Evaluation regarding devices suspected to presenting an unacceptable risk or non-compliance *For New Products: New technology and not line extension TÜV SÜD Product Service Slide 36


37 Clinical Evaluation according to MEDDEV Rev 3 Which route should you follow for the device under consideration? Equivalence Route TÜV SÜD Product Service Clinical Investigation Route Slide 37
38 EU Regulations Proposal Clinical Evaluation Article 49: Clinical Evaluation Manufacturers shall conduct a clinical evaluation in accordance with the principles set out in this Article and Part A of Annex XIII. A clinical evaluation shall follow a defined and methodologically sound procedure based on either of the following: A critical evaluation of the relevant scientific literature A critical evaluation of the results of all clinical i l investigations A critical evaluation of the combined clinical data TÜV SÜD Product Service Slide 38
39 EU Regulations Proposal Clinical Investigations Requirements For Class III medical devices and Implantable medical devices clinical investigations shall be performed unless it is duly justified to rely on exisiting clinical data alone Demonstration of equivalence shall generally not be considered as sufficient justification within the meaning of this sentence Equivalence can only be demonstrated when the device has the same intended purpose and when the technical and biological characteristics of the devices and the medical procedures applied are similar il to such an extent t thatt there would be not a clinically significant difference in the safety and performance of the devices. Preamble (47) in the proposed regulation Clinical investigations in line with major international guidance in this field, such as the international standard ISO 14155:2011. TÜV SÜD Product Service Slide 39




40 EU Regulations Proposal Clinical Investigations Requirements Council In the case of implantable devices and devices falling within class III, clinical investigations shall be performed except if the device has been designed by modifications of a device already marketed by the same manufacturer if the modifications have been scientifically demonstrated by the manufacturer and accepted by the Notified Body as being equivalent. In this case the Notified Body shall check that the PMCF plan is appropriate and includes post market studies to demonstrate the safety and performance of the device. *Clear contract between different device manufacturer TÜV SÜD Product Service Slide 40
41 EU Regulations Proposal PMCF Requirements A PMCF Study To be performed according to a PMCF Plan with the aim of: confirming the safety and performance of the device throughout its expected lifetime identifying previously unknown side-effects and monitoring the identified side effects/contra-indications identifying and analyzing emergent risks on the basis of factual evidence assuring the continued acceptability of the benefit/risk ratio identifying possible systematic misuse or off-label use of the device TÜV SÜD Product Service Slide 41
42 EU Regulations Proposal PMCF Requirements Council Proposal For devices classified as class III and implantable devices, the PMCF report, and if indicated, the summary of safety and clinical performance referred to in Article 26(1) shall be updated at least annually with ih these data. TÜV SÜD Product Service Slide 42









43 Questions? Dr. Bassil Akra Director cce For enquiries, me at: Global l website: di i Stay informed and updated with our Healthcare & Medical Device newsletter: TÜV TÜV SÜD SÜD Product Service Lighting Services Slide 43
Requirements on clinical data in Europe
 Requirements on clinical data in Europe Dr. Bassil Akra Director Global Clinical Affairs TÜV SÜD Product Service Current Medical Device Directives Applicable Directives Active Implantable Medical Devices
Requirements on clinical data in Europe Dr. Bassil Akra Director Global Clinical Affairs TÜV SÜD Product Service Current Medical Device Directives Applicable Directives Active Implantable Medical Devices
WEBINAR on the new Medical Device Regulation One-stop testing, inspection, certification and training solutions. TÜV SÜD Product Service
 WEBINAR on the new Medical Device Regulation 2017-07-12 One-stop testing, inspection, certification and training solutions TÜV SÜD Product Service TÜV SÜD at a glance 150+ YEARS OF QUALITY, SAFETY & SUSTAINABILITY
WEBINAR on the new Medical Device Regulation 2017-07-12 One-stop testing, inspection, certification and training solutions TÜV SÜD Product Service TÜV SÜD at a glance 150+ YEARS OF QUALITY, SAFETY & SUSTAINABILITY
Mapping Your Success 2013 BSI Healthcare Roadshow: Mobile Health Software, Mobile Phones and Telemedicine Under the EU Approach
 Mapping Your Success 2013 BSI Healthcare Roadshow: Mobile Health Software, Mobile Phones and Telemedicine Under the EU Approach Telemedicine & mhealth Definition of Telemedicine: Use of telecommunication
Mapping Your Success 2013 BSI Healthcare Roadshow: Mobile Health Software, Mobile Phones and Telemedicine Under the EU Approach Telemedicine & mhealth Definition of Telemedicine: Use of telecommunication
Regulatory Aspects of Digital Healthcare Solutions
 Regulatory Aspects of Digital Healthcare Solutions TÜV SÜD Product Service GmbH Dr. Markus Siebert Rev. 02 / 2017 02.05.2017 TÜV SÜD Product Service GmbH Slide 1 Contents Digital solutions as Medical Device
Regulatory Aspects of Digital Healthcare Solutions TÜV SÜD Product Service GmbH Dr. Markus Siebert Rev. 02 / 2017 02.05.2017 TÜV SÜD Product Service GmbH Slide 1 Contents Digital solutions as Medical Device
Med-Info. Council Directive 93/42/EEC on medical devices. TÜV SÜD Product Service GmbH
 Med-Info International expert information for the medical device industry Council Directive 93/42/E on medical devices Practice-oriented summary of the most important aspects and requirements contained
Med-Info International expert information for the medical device industry Council Directive 93/42/E on medical devices Practice-oriented summary of the most important aspects and requirements contained
Med-Info. Council Directive 93/42/EEC on Medical Devices. TÜV SÜD Product Service GmbH
 Med-Info International expert information for the Medical Device industry Council Directive 93/42/E on Medical Devices Practice-oriented summary of the most important aspects and requirements contained
Med-Info International expert information for the Medical Device industry Council Directive 93/42/E on Medical Devices Practice-oriented summary of the most important aspects and requirements contained
Med-Info. Council Directive 93/42/EEC on medical devices. TÜV SÜD Product Service GmbH
 Med-Info International expert information for the medical device industry Council Directive 93/42/E on medical devices Practice-oriented summary of the most important aspects and requirements contained
Med-Info International expert information for the medical device industry Council Directive 93/42/E on medical devices Practice-oriented summary of the most important aspects and requirements contained
Submission of information in the public consultation on potential candidates for substitution under the Biocidal Products Regulation
 Submission of information in the public consultation on potential candidates for substitution under the Biocidal Products Regulation Version 3.0 May 2017 2 Submission of information in the public consultation
Submission of information in the public consultation on potential candidates for substitution under the Biocidal Products Regulation Version 3.0 May 2017 2 Submission of information in the public consultation
ENCePP Code of Conduct Implementation Guidance for Sharing of ENCePP Study Data
 EMA/409316/2010 Revision 2, dated 14 July 2016 European Network of Centres for Pharmacoepidemiology and Pharmacovigilance ENCePP Code of Conduct Implementation Guidance for Sharing of ENCePP Study Data
EMA/409316/2010 Revision 2, dated 14 July 2016 European Network of Centres for Pharmacoepidemiology and Pharmacovigilance ENCePP Code of Conduct Implementation Guidance for Sharing of ENCePP Study Data
Medical Device Usability
 Medical Device Usability David Adams Global Head, Active Medical Devices Add logo on slide 4 here Topics What is usability? Why usability is so important The regulatory requirements EN 62366 Usability
Medical Device Usability David Adams Global Head, Active Medical Devices Add logo on slide 4 here Topics What is usability? Why usability is so important The regulatory requirements EN 62366 Usability
LNE/G-MED North America, Inc
 LNE/G-MED North America, Inc Medical Device Usability: Highlights of European Regulations and the Latest Standards Do not distribute or reproduce without permission 1 Sara Jafari, Ph.D., Medical Device
LNE/G-MED North America, Inc Medical Device Usability: Highlights of European Regulations and the Latest Standards Do not distribute or reproduce without permission 1 Sara Jafari, Ph.D., Medical Device
IVDR Breakout. Copyright 2017 BSI. All rights reserved.
 IVDR Breakout 1 IVDR Classification and conformity assessment 2 Classification- IVDR 3 Classification of IVDs Re-classification of IVDs will mean 80-90 % will no longer be able to self certify conformity
IVDR Breakout 1 IVDR Classification and conformity assessment 2 Classification- IVDR 3 Classification of IVDs Re-classification of IVDs will mean 80-90 % will no longer be able to self certify conformity
EA Document for Recognition of Verifiers under the EU ETS Directive
 Publication Reference EA-6/03: 2010 Mandatory Document EA Document for Recognition of Verifiers under the EU ETS Directive PURPOSE This document has been prepared by a working group under the direction
Publication Reference EA-6/03: 2010 Mandatory Document EA Document for Recognition of Verifiers under the EU ETS Directive PURPOSE This document has been prepared by a working group under the direction
Guidance for the format and content of the final study report of non-interventional post-authorisation safety studies
 23 January 2013 EMA/48663/2013 Patient Health Protection Guidance for the format and content of the final study report of non-interventional post-authorisation safety studies Introduction From 10 January
23 January 2013 EMA/48663/2013 Patient Health Protection Guidance for the format and content of the final study report of non-interventional post-authorisation safety studies Introduction From 10 January
!"# $ # # $ $ % $ &% $ '"# $ ()&*&)+(( )+(( )
&*&)+(( )+(( )") !"# # # % &% '"# ) !#, ' "# " "# -. / # 0 0 0 0 0 "0 "# " # 1 #! " " 0 0 0 0 0 0 2# 0 # # 3 ' 4 56 7-56 87 9# 5 6 7 6 & 0 " : 9 ; 4 " #! 0 - '% # % "# " "# " < 4 "! % " % 4 % % 9# 4 56 87 = 4 > 0 " %!#
!"# # # % &% '"# ) !#, ' "# " "# -. / # 0 0 0 0 0 "0 "# " # 1 #! " " 0 0 0 0 0 0 2# 0 # # 3 ' 4 56 7-56 87 9# 5 6 7 6 & 0 " : 9 ; 4 " #! 0 - '% # % "# " "# " < 4 "! % " % 4 % % 9# 4 56 87 = 4 > 0 " %!#
Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679
 Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679 Adopted on 25 May 2018 Contents 1. Introduction... 2 1.1. Scope
Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679 Adopted on 25 May 2018 Contents 1. Introduction... 2 1.1. Scope
UDI in Europe. Mr. Salvatore Scalzo, Policy and Legal Officer, Medical Devices, DG GROW, European Commission. 19 October 2017
 UDI in Europe Mr. Salvatore Scalzo, Policy and Legal Officer, Medical Devices, DG GROW, European Commission 19 October 2017 The new EU Medical Device Regulations: Introduction to the future EU UDI System
UDI in Europe Mr. Salvatore Scalzo, Policy and Legal Officer, Medical Devices, DG GROW, European Commission 19 October 2017 The new EU Medical Device Regulations: Introduction to the future EU UDI System
COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) GUIDELINE ON REQUIREMENTS FOR PLASMA MASTER FILE (PMF) CERTIFICATION
 GUIDELINE ON REQUIREMENTS FOR PLASMA MASTER FILE (PMF) CERTIFICATION") The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 26 February 2004 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) GUIDELINE ON REQUIREMENTS
The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 26 February 2004 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) GUIDELINE ON REQUIREMENTS
ACCREDITATION: A BRIEFING FOR GOVERNMENTS AND REGULATORS
 ACCREDITATION: A BRIEFING FOR GOVERNMENTS AND REGULATORS Accreditation is continuously gaining recognition as an important technical tool in the delivery of objectives across an increasing range of policy
ACCREDITATION: A BRIEFING FOR GOVERNMENTS AND REGULATORS Accreditation is continuously gaining recognition as an important technical tool in the delivery of objectives across an increasing range of policy
AUDITOR / LEAD AUDITOR PHARMACEUTICAL AND MEDICAL DEVICE INDUSTRY
 Requirement specification Certification of individuals: AUDITOR / LEAD AUDITOR PHARMACEUTICAL AND MEDICAL DEVICE INDUSTRY Requirement specification Auditor Lead Auditor rev 5.docx Page 1 1 Introduction
Requirement specification Certification of individuals: AUDITOR / LEAD AUDITOR PHARMACEUTICAL AND MEDICAL DEVICE INDUSTRY Requirement specification Auditor Lead Auditor rev 5.docx Page 1 1 Introduction
Content of mandatory certificates
 Chapter: 2.5.1 Conformity assessment procedures; General rules Text:... Key words: certificate, certificate of competence, 1. Purpose The purpose of this recommendation is to provide guidance on the minimum
Chapter: 2.5.1 Conformity assessment procedures; General rules Text:... Key words: certificate, certificate of competence, 1. Purpose The purpose of this recommendation is to provide guidance on the minimum
Med-Info. Malaysia Medical Device Regulations. TÜV SÜD Product Service GmbH. International expert information for the medical device industry
 Med-Info International expert information for the medical device industry Malaysia Medical Device Regulations Passed in 2012, the Medical Device Act (Act 737) and the Medical Device Authority Act 2012
Med-Info International expert information for the medical device industry Malaysia Medical Device Regulations Passed in 2012, the Medical Device Act (Act 737) and the Medical Device Authority Act 2012
Data Processing Clauses
 Data Processing Clauses The examples of processing clauses below are proposed pending the adoption of standard contractual clauses within the meaning of Article 28.8 of general data protection regulation.
Data Processing Clauses The examples of processing clauses below are proposed pending the adoption of standard contractual clauses within the meaning of Article 28.8 of general data protection regulation.
Guidelines 4/2018 on the accreditation of certification bodies under Article 43 of the General Data Protection Regulation (2016/679)
") Guidelines 4/2018 on the accreditation of certification bodies under Article 43 of the General Data Protection Regulation (2016/679) Adopted on 4 December 2018 Adopted 1 Contents 1 Introduction... 3 2
Guidelines 4/2018 on the accreditation of certification bodies under Article 43 of the General Data Protection Regulation (2016/679) Adopted on 4 December 2018 Adopted 1 Contents 1 Introduction... 3 2
QP Current Practices, Challenges and Mysteries. Caitriona Lenagh 16 March 2012
 QP Current Practices, Challenges and Mysteries Caitriona Lenagh 16 March 2012 Agenda QP Roles and Responsibilities QP Current Practices Supply Chain Verification Study Specific Information Lot Specific
QP Current Practices, Challenges and Mysteries Caitriona Lenagh 16 March 2012 Agenda QP Roles and Responsibilities QP Current Practices Supply Chain Verification Study Specific Information Lot Specific
Rules for LNE Certification of Management Systems
 Rules for LNE Certification of Management Systems Application date: March 10 th, 2017 Rev. 040716 RULES FOR LNE CERTIFICATION OF MANAGEMENT SYSTEMS CONTENTS 1. PURPOSE... 3 2. SCOPE... 3 3. DEFINITION
Rules for LNE Certification of Management Systems Application date: March 10 th, 2017 Rev. 040716 RULES FOR LNE CERTIFICATION OF MANAGEMENT SYSTEMS CONTENTS 1. PURPOSE... 3 2. SCOPE... 3 3. DEFINITION
EA-7/05 - EA Guidance on the Application of ISO/IEC 17021:2006 for Combined Audits
 Publication Reference EA-7/05 EA Guidance on the Application of ISO/IEC 17021:2006 for Combined Audits PURPOSE This document has been prepared by a task force under the direction of the European Cooperation
Publication Reference EA-7/05 EA Guidance on the Application of ISO/IEC 17021:2006 for Combined Audits PURPOSE This document has been prepared by a task force under the direction of the European Cooperation
UDI Implementation Update. GS1 UK Healthcare Conference - 22 November 2017 John Wilkinson OBE Medicines and Healthcare Products Regulatory Agency
 UDI Implementation Update GS1 UK Healthcare Conference - 22 November 2017 John Wilkinson OBE Medicines and Healthcare Products Regulatory Agency 2 Why new European medical device and IVD regulations? Old
UDI Implementation Update GS1 UK Healthcare Conference - 22 November 2017 John Wilkinson OBE Medicines and Healthcare Products Regulatory Agency 2 Why new European medical device and IVD regulations? Old
The Accreditation and Verification Regulation - Verification report
 EUROPEAN COMMISSION DIRECTORATE-GENERAL CLIMATE ACTION Directorate A - International and Climate Strategy CLIMA.A.3 - Monitoring, Reporting, Verification Guidance Document The Accreditation and Verification
EUROPEAN COMMISSION DIRECTORATE-GENERAL CLIMATE ACTION Directorate A - International and Climate Strategy CLIMA.A.3 - Monitoring, Reporting, Verification Guidance Document The Accreditation and Verification
APPROVAL SHEET PROCEDURE INFORMATION SECURITY MANAGEMENT SYSTEM CERTIFICATION. PT. TÜV NORD Indonesia PS - TNI 001 Rev.05
 APPROVAL SHEET PROCEDURE INFORMATION SECURITY MANAGEMENT SYSTEM CERTIFICATION PT. TÜV NORD Indonesia PS - TNI 001 Rev.05 Created : 20-06-2016 Checked: 20-06-2016 Approved : 20-06-2016 Indah Lestari Karlina
APPROVAL SHEET PROCEDURE INFORMATION SECURITY MANAGEMENT SYSTEM CERTIFICATION PT. TÜV NORD Indonesia PS - TNI 001 Rev.05 Created : 20-06-2016 Checked: 20-06-2016 Approved : 20-06-2016 Indah Lestari Karlina
ARTICLE 29 DATA PROTECTION WORKING PARTY
 ARTICLE 29 DATA PROTECTION WORKING PARTY 18/EN WP261 Article 29 Working Party Draft Guidelines on the accreditation of certification bodies under Regulation (EU) 2016/679 Adopted on 6 february 2018 1 THE
ARTICLE 29 DATA PROTECTION WORKING PARTY 18/EN WP261 Article 29 Working Party Draft Guidelines on the accreditation of certification bodies under Regulation (EU) 2016/679 Adopted on 6 february 2018 1 THE
Scheme Document. For more information or help with your application contact BRE Global on +44 (0) or
 or") Page: Page 1 of 15 1. Introduction This certification scheme has been designed to promote sustainable production of construction products and materials. Responsible sourcing includes organisational management,
Page: Page 1 of 15 1. Introduction This certification scheme has been designed to promote sustainable production of construction products and materials. Responsible sourcing includes organisational management,
Voluntary certification at an intermediate stage of manufacture
 Chapter: 2.15 Other Text: Key words: MDD results of any assessment and verification operation, which where appropriate have been carried out in accordance with this Directive at an intermediate stage of
Chapter: 2.15 Other Text: Key words: MDD results of any assessment and verification operation, which where appropriate have been carried out in accordance with this Directive at an intermediate stage of
Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679
 Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679 Adopted on 23 January 2019 1 Table of contents 1.1 Scope of the
Guidelines 1/2018 on certification and identifying certification criteria in accordance with Articles 42 and 43 of the Regulation 2016/679 Adopted on 23 January 2019 1 Table of contents 1.1 Scope of the
COMMISSION IMPLEMENTING DECISION (EU)
") L 127/32 18.5.2016 COMMISSION IMPLEMTING DECISION (EU) 2016/770 of 14 April 2016 establishing a common format for the submission of information concerning the operation of the procedures pursuant to Regulation
L 127/32 18.5.2016 COMMISSION IMPLEMTING DECISION (EU) 2016/770 of 14 April 2016 establishing a common format for the submission of information concerning the operation of the procedures pursuant to Regulation
1. STRATEGIC PLANNING
 RAC (EU) EXAMINATION SUBJECTS & FORMAT The European RAC Examination is a knowledge-based examination addressing European Union laws, regulations, policies and guidelines affecting medical RAC devices,
RAC (EU) EXAMINATION SUBJECTS & FORMAT The European RAC Examination is a knowledge-based examination addressing European Union laws, regulations, policies and guidelines affecting medical RAC devices,
BRE Global Limited Scheme Document SD 186: Issue No December 2017
 BRE Global Limited Scheme Document SD 186: Issue No. 11.1 Commercial-in-Confidence Page 1 of 9 Introduction This certification scheme has been designed to promote the use of sustainable materials and
BRE Global Limited Scheme Document SD 186: Issue No. 11.1 Commercial-in-Confidence Page 1 of 9 Introduction This certification scheme has been designed to promote the use of sustainable materials and
BEST PRACTICE GUIDE for The classification of unforeseen variations
 EMA/CMDv/499821/2008 CMDv/BPG/015 BEST PRACTICE GUIDE for The classification of unforeseen variations Edition number: 02 Edition date: 18 December 2012 Implementation date: 18 December 2012 1 1 Amending
EMA/CMDv/499821/2008 CMDv/BPG/015 BEST PRACTICE GUIDE for The classification of unforeseen variations Edition number: 02 Edition date: 18 December 2012 Implementation date: 18 December 2012 1 1 Amending
GMO Register User Guide
 GMO Register User Guide A. Rana and F. Foscarini Institute for Health and Consumer Protection 2007 EUR 22697 EN The mission of the Institute for Health and Consumer Protection is to provide scientific
GMO Register User Guide A. Rana and F. Foscarini Institute for Health and Consumer Protection 2007 EUR 22697 EN The mission of the Institute for Health and Consumer Protection is to provide scientific
CEN and CENELEC Position Paper on the draft regulation ''Cybersecurity Act''
 CEN Identification number in the EC register: 63623305522-13 CENELEC Identification number in the EC register: 58258552517-56 CEN and CENELEC Position Paper on the draft regulation ''Cybersecurity Act''
CEN Identification number in the EC register: 63623305522-13 CENELEC Identification number in the EC register: 58258552517-56 CEN and CENELEC Position Paper on the draft regulation ''Cybersecurity Act''
SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF INFOR- MATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001)
") BELAC 2-405-ISMS R0 2017 SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF INFOR- MATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001) The only valid versions of the documents
BELAC 2-405-ISMS R0 2017 SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF INFOR- MATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001) The only valid versions of the documents
SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF FOOD SAFETY MANAGEMENT SYSTEMS
 BELAC 2-405-FSMS Rev 1-2017 SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF FOOD SAFETY MANAGEMENT SYSTEMS The only valid versions of the documents of the BELAC management
BELAC 2-405-FSMS Rev 1-2017 SPECIFIC PROVISIONS FOR THE ACCREDITATION OF CERTIFICATION BODIES IN THE FIELD OF FOOD SAFETY MANAGEMENT SYSTEMS The only valid versions of the documents of the BELAC management
TURKISH STANDARDS INSTITUTION
 TURKISH STANDARDS INSTITUTION NEW GLOBAL OLD APPROACH REGULATIONS CONFORMITY ASSESSMENT PROCEDURES AND PRINCIPLES Board Decision : 29.04.2014 Date 50.-236 Validity date : 05.05.2014 CHAPTER ONE Objective,
TURKISH STANDARDS INSTITUTION NEW GLOBAL OLD APPROACH REGULATIONS CONFORMITY ASSESSMENT PROCEDURES AND PRINCIPLES Board Decision : 29.04.2014 Date 50.-236 Validity date : 05.05.2014 CHAPTER ONE Objective,
National Accreditation Scheme
 National Accreditation Scheme Rules of Procedure on the Preparation of Accreditation Audit Cycle NAR-25 Edition 3 Version 1 Approved by: Csaba Bodroghelyi Deputy Director General Responsible for preparation:
National Accreditation Scheme Rules of Procedure on the Preparation of Accreditation Audit Cycle NAR-25 Edition 3 Version 1 Approved by: Csaba Bodroghelyi Deputy Director General Responsible for preparation:
Conformity assessment
 Training Course on Conformity and Interoperability, Tunis-Tunisia, from 22 to 26 May 2017 Conformity assessment Presented by: Karim Loukil & Kaïs Siala Page 1 Today s Objectives Present basic information
Training Course on Conformity and Interoperability, Tunis-Tunisia, from 22 to 26 May 2017 Conformity assessment Presented by: Karim Loukil & Kaïs Siala Page 1 Today s Objectives Present basic information
Solutions that ensure safety, reliability and compliance.
 RED Transition and Implementation by TÜV SÜD Radu Gosav, Senior Manager EMC&RF, Greater China Solutions that ensure safety, reliability and compliance. TÜV SÜD Greater China Slide 1 17-02-22 1 RED Introduction
RED Transition and Implementation by TÜV SÜD Radu Gosav, Senior Manager EMC&RF, Greater China Solutions that ensure safety, reliability and compliance. TÜV SÜD Greater China Slide 1 17-02-22 1 RED Introduction
INTERNATIONAL TELECOMMUNICATION UNION
 INTERNATIONAL TELECOMMUNICATION UNION ITU-T E.212 TELECOMMUNICATION STANDARDIZATION SECTOR OF ITU (05/2004) SERIES E: OVERALL NETWORK OPERATION, TELEPHONE SERVICE, SERVICE OPERATION AND HUMAN FACTORS International
INTERNATIONAL TELECOMMUNICATION UNION ITU-T E.212 TELECOMMUNICATION STANDARDIZATION SECTOR OF ITU (05/2004) SERIES E: OVERALL NETWORK OPERATION, TELEPHONE SERVICE, SERVICE OPERATION AND HUMAN FACTORS International
Base Standard Program ISO Medical Device CB Application for Accreditation
 Base Standard Program ISO 13485 Medical Device CB Application for Accreditation FA 5006 Authority: Accreditation Manager Effective: 2016/11/11 Section 1: CB Name, Contact Information, and Processing Fees
Base Standard Program ISO 13485 Medical Device CB Application for Accreditation FA 5006 Authority: Accreditation Manager Effective: 2016/11/11 Section 1: CB Name, Contact Information, and Processing Fees
Standard Operating Procedure. SOP full title: Sponsor processes for reporting Suspected Unexpected Serious Adverse Reactions
 Standard Operating Procedure SOP number: SOP full title: SOP-JRO-03-003 Sponsor processes for reporting Suspected Unexpected Serious Adverse Reactions SOP effective: 23/05/2017 Review date: 23/05/2019
Standard Operating Procedure SOP number: SOP full title: SOP-JRO-03-003 Sponsor processes for reporting Suspected Unexpected Serious Adverse Reactions SOP effective: 23/05/2017 Review date: 23/05/2019
KENYA ACCREDITATION SERVICE
 KENAS-GUD-010 01 22/06/2013 22/07/2013 GUD 1 of 9 Approval and Authorisation Completion of the following signature blocks signifies the review and approval of this Document. Name Job Title / Role Signature
KENAS-GUD-010 01 22/06/2013 22/07/2013 GUD 1 of 9 Approval and Authorisation Completion of the following signature blocks signifies the review and approval of this Document. Name Job Title / Role Signature
GUIDANCE AND INTERPRETATION DOCUMENTS TO THE REQUIREMENTS FOR THE COMPETENCE OF CONFORMITY ASSESSMENT BODIES
 GUIDANCE AND INTERPRETATION DOCUMENTS TO THE REQUIREMENTS FOR THE COMPETENCE OF CONFORMITY ASSESSMENT BODIES Table of Contents 1 PURPOSE... 2 2 GENERAL... 2 3 GUIDANCE AND INTERPRETATIVE DOCUMENTS... 2
GUIDANCE AND INTERPRETATION DOCUMENTS TO THE REQUIREMENTS FOR THE COMPETENCE OF CONFORMITY ASSESSMENT BODIES Table of Contents 1 PURPOSE... 2 2 GENERAL... 2 3 GUIDANCE AND INTERPRETATIVE DOCUMENTS... 2
SmPC and. & New QRD. Spanish Medicines Agency (AEMPS)
") SmPC and Labelling User Testing & New QRD template Blanca García-Ochoa Martín Spanish Medicines Agency (AEMPS) Legal framework Directive 2001/83/EC of the European Parliament and of the Council of 6 November
SmPC and Labelling User Testing & New QRD template Blanca García-Ochoa Martín Spanish Medicines Agency (AEMPS) Legal framework Directive 2001/83/EC of the European Parliament and of the Council of 6 November
IAF Mandatory Document KNOWLEDGE REQUIREMENTS FOR ACCREDITATION BODY PERSONNEL FOR INFORMATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001)
") IAF Mandatory Document KNOWLEDGE REQUIREMENTS FOR ACCREDITATION BODY PERSONNEL FOR INFORMATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001) (IAF MD 13:2015) Issue 1 IAF MD - Knowledge Requirements for Accreditation
IAF Mandatory Document KNOWLEDGE REQUIREMENTS FOR ACCREDITATION BODY PERSONNEL FOR INFORMATION SECURITY MANAGEMENT SYSTEMS (ISO/IEC 27001) (IAF MD 13:2015) Issue 1 IAF MD - Knowledge Requirements for Accreditation
INSPIRE status report
 INSPIRE Team INSPIRE Status report 29/10/2010 Page 1 of 7 INSPIRE status report Table of contents 1 INTRODUCTION... 1 2 INSPIRE STATUS... 2 2.1 BACKGROUND AND RATIONAL... 2 2.2 STAKEHOLDER PARTICIPATION...
INSPIRE Team INSPIRE Status report 29/10/2010 Page 1 of 7 INSPIRE status report Table of contents 1 INTRODUCTION... 1 2 INSPIRE STATUS... 2 2.1 BACKGROUND AND RATIONAL... 2 2.2 STAKEHOLDER PARTICIPATION...
Global Specification Protocol for Organisations Certifying to an ISO Standard related to Market, Opinion and Social Research.
 CONTENTS i. INTRODUCTION 3 ii. OVERVIEW SPECIFICATION PROTOCOL DOCUMENT DEVELOPMENT PROCESS 4 1. SCOPE 5 2. DEFINITIONS 5 3. REFERENCES 6 4. MANAGEMENT STANDARDS FOR APPROVED CERTIFICATION BODIES 6 4.1
CONTENTS i. INTRODUCTION 3 ii. OVERVIEW SPECIFICATION PROTOCOL DOCUMENT DEVELOPMENT PROCESS 4 1. SCOPE 5 2. DEFINITIONS 5 3. REFERENCES 6 4. MANAGEMENT STANDARDS FOR APPROVED CERTIFICATION BODIES 6 4.1
ANNEX ANNEX. to the COMMISSION IMPLEMENTING REGULATION (EU)
") Ref. Ares(2018)1944240-11/04/2018 EUROPEAN COMMISSION Brussels, XXX [ ](2018) XXX draft ANNEX ANNEX to the COMMISSION IMPLEMENTING REGULATION (EU) laying down minimum requirements implementing the provisions
Ref. Ares(2018)1944240-11/04/2018 EUROPEAN COMMISSION Brussels, XXX [ ](2018) XXX draft ANNEX ANNEX to the COMMISSION IMPLEMENTING REGULATION (EU) laying down minimum requirements implementing the provisions
UDI in the MDR. Economic Operators The new regulations create Economic Operators who play a role in the UDI system.
 UDI in the MDR The European Union intends to replace the existing directives related to medical device, Active Implantable Medical Devices, In Vitro Diagnostic Devices, and Medical Devices, with two regulations.
UDI in the MDR The European Union intends to replace the existing directives related to medical device, Active Implantable Medical Devices, In Vitro Diagnostic Devices, and Medical Devices, with two regulations.
European Aviation Safety Agency
 European Aviation Safety Agency EASA Management Board Decision 12-2007 Amending the products certification procedure MB meeting 04-2007 (11 September 2007) DECISION OF THE MANAGEMENT BOARD AMENDING DECISION
European Aviation Safety Agency EASA Management Board Decision 12-2007 Amending the products certification procedure MB meeting 04-2007 (11 September 2007) DECISION OF THE MANAGEMENT BOARD AMENDING DECISION
European Medical Device Regulations: Keys to Success in this Era of Increased Medical Device Oversight
 European Medical Device Regulations: Keys to Success in this Era of Increased Medical Device Oversight American Medical Device Summit Chicago, IL September 21-22, 2015 Richard DeRisio, M.S. RAC Mark Twain,
European Medical Device Regulations: Keys to Success in this Era of Increased Medical Device Oversight American Medical Device Summit Chicago, IL September 21-22, 2015 Richard DeRisio, M.S. RAC Mark Twain,
ILNAS/PSCQ/Pr004 Qualification of technical assessors
 Version 1.1 21.6.2016 Page 1 of 6 ILNAS/PSCQ/Pr004 Qualification of technical assessors Modifications: review of the document 1, avenue du Swing L-4367 Belvaux Tél.: (+352) 247 743-53 Fax: (+352) 247 943-50
Version 1.1 21.6.2016 Page 1 of 6 ILNAS/PSCQ/Pr004 Qualification of technical assessors Modifications: review of the document 1, avenue du Swing L-4367 Belvaux Tél.: (+352) 247 743-53 Fax: (+352) 247 943-50
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL New Approach Industries, Tourism and CSR Construction, Pressure Equipment, Metrology Brussels, 21 st December 2009 M/457 EN Standardisation
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL New Approach Industries, Tourism and CSR Construction, Pressure Equipment, Metrology Brussels, 21 st December 2009 M/457 EN Standardisation
The Accreditation and Verification Regulation - Sampling
 EUROPEAN COMMISSION DIRECTORATE-GENERAL CLIMATE ACTION Directorate C - Climate Strategy, Governance and Emissions from non-trading sectors CLIMA.C.2 - Governance and Effort Sharing Guidance Document The
EUROPEAN COMMISSION DIRECTORATE-GENERAL CLIMATE ACTION Directorate C - Climate Strategy, Governance and Emissions from non-trading sectors CLIMA.C.2 - Governance and Effort Sharing Guidance Document The
USING STANDARDS TO ASSESS THE COMPETENCE OF CONFORMITY
 Ref. Ares(2014)2675967-13/08/2014 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Regulaty policy Regulaty Approach f the free movement of goods NOTE TO THE SENIOR OFFICIALS GROUP ON STANDARDISATION
Ref. Ares(2014)2675967-13/08/2014 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Regulaty policy Regulaty Approach f the free movement of goods NOTE TO THE SENIOR OFFICIALS GROUP ON STANDARDISATION
ECC Recommendation (17)04. Numbering for ecall
04. Numbering for ecall") ECC Recommendation (17)04 Numbering for ecall Approved 22 November 2017 ECC/REC/(17)04 Page 2 INTRODUCTION ecall is a service designed for automotive vehicles to provide quick emergency response in case
ECC Recommendation (17)04 Numbering for ecall Approved 22 November 2017 ECC/REC/(17)04 Page 2 INTRODUCTION ecall is a service designed for automotive vehicles to provide quick emergency response in case
ISO/IEC INTERNATIONAL STANDARD
 INTERNATIONAL STANDARD ISO/IEC 27006 Second edition 2011-12-01 Information technology Security techniques Requirements for bodies providing audit and certification of information security management systems
INTERNATIONAL STANDARD ISO/IEC 27006 Second edition 2011-12-01 Information technology Security techniques Requirements for bodies providing audit and certification of information security management systems
Sources of Test Reports for TUV SUD BABT Product Certification BABT766. TUV SUD BABT is a certification body of. TUV SUD BABT 2015 Issue 7
 BABT766 Sources of Test Reports for TUV SUD BABT Product Certification TUV SUD BABT is a certification body of TUV SUD BABT 2015 Page 1 of 5 Contents 1 Introduction... 2 2 Background... 2 3 General...
BABT766 Sources of Test Reports for TUV SUD BABT Product Certification TUV SUD BABT is a certification body of TUV SUD BABT 2015 Page 1 of 5 Contents 1 Introduction... 2 2 Background... 2 3 General...
Vaccine data collection tool Oct Functions, Indicators & Sub-Indicators
 data collection tool Oct. 2011 A. National Regulatory System RS01: Legal framework for establishment of a regulatory system, mandate and enforcement power for each function RS01.01: Legislation or and
data collection tool Oct. 2011 A. National Regulatory System RS01: Legal framework for establishment of a regulatory system, mandate and enforcement power for each function RS01.01: Legislation or and
Isabella Marta, Head of Marketing Authorization Department Italian Medicines Agency
 API Regulatory Compliance: GMP Inspections and Marketing Authorization Isabella Marta, Head of Marketing Authorization Department Italian Medicines Agency The Place of the Certification procedure in the
API Regulatory Compliance: GMP Inspections and Marketing Authorization Isabella Marta, Head of Marketing Authorization Department Italian Medicines Agency The Place of the Certification procedure in the
BEST PRACTICE GUIDE for Type IB Variations. CMD(v)/BPG/005. Edition 00. Edition date: Implementation date:
/BPG/005. Edition 00. Edition date: Implementation date:") Co-ordination Group for Mutual Recognition and Decentralised Procedures - Veterinary EMEA/CMD(v)/115405/2005 BEST PRACTICE GUIDE for Type IB Variations Edition 00 Edition date: 10-12-2005 Implementation
Co-ordination Group for Mutual Recognition and Decentralised Procedures - Veterinary EMEA/CMD(v)/115405/2005 BEST PRACTICE GUIDE for Type IB Variations Edition 00 Edition date: 10-12-2005 Implementation
How the European Commission is supporting innovation in mobile health technologies Nordic Mobile Healthcare Technology Congress 2015
 How the European Commission is supporting innovation in mobile health technologies Nordic Mobile Healthcare Technology Congress 2015 Claudia Prettner, Unit for Health and Well-Being, DG CONNECT Table of
How the European Commission is supporting innovation in mobile health technologies Nordic Mobile Healthcare Technology Congress 2015 Claudia Prettner, Unit for Health and Well-Being, DG CONNECT Table of
Inspection and Certification for Individual Farms, Smallholder Group Certification S S R A N A S R S C I E N T I S T
 Inspection and Certification for Individual Farms, Smallholder Group Certification S S R A N A S R S C I E N T I S T What is Certification? Organic certification system is a quality assurance initiative,
Inspection and Certification for Individual Farms, Smallholder Group Certification S S R A N A S R S C I E N T I S T What is Certification? Organic certification system is a quality assurance initiative,
CONTINUOUS PROFESSIONAL DEVELOPMENT (CPD) POLICY
 POLICY") CONTINUOUS PROFESSIONAL DEVELOPMENT (CPD) POLICY SUMMARY: This defined as a framework that encourages continuous updating of professional knowledge, personal skills and competencies. DATE OF APPROVAL FOR
CONTINUOUS PROFESSIONAL DEVELOPMENT (CPD) POLICY SUMMARY: This defined as a framework that encourages continuous updating of professional knowledge, personal skills and competencies. DATE OF APPROVAL FOR
INAB Mandatory and Guidance Documents Policy and Index
 INAB Mandatory and Guidance s Policy and Index This publication is aimed at assisting in determining what documents are relevant to various organisations and at providing contact points for accessing such
INAB Mandatory and Guidance s Policy and Index This publication is aimed at assisting in determining what documents are relevant to various organisations and at providing contact points for accessing such
Frequently Asked Questions
 Frequently Asked Questions ISO 15189 Accreditation Program cap.org Contents ISO and the International Organization for Standardization What does ISO stand for? (page 3) What is the International Organization
Frequently Asked Questions ISO 15189 Accreditation Program cap.org Contents ISO and the International Organization for Standardization What does ISO stand for? (page 3) What is the International Organization
INAB Mandatory and Guidance Documents Policy and Index
 INAB Mandatory and Guidance s Policy and Index This publication is aimed at assisting in determining what documents are relevant to various organisations and at providing contact points for accessing such
INAB Mandatory and Guidance s Policy and Index This publication is aimed at assisting in determining what documents are relevant to various organisations and at providing contact points for accessing such
UK EPR GDA PROJECT. Name/Initials Date 30/06/2011 Name/Initials Date 30/06/2011. Resolution Plan Revision History
 RP unique number: GI-UKEPR-CI-01-RP 0 30/06/2011 1 of 19 Approved for EDF by: A. PETIT Approved for AREVA by: C. WOOLDRIDGE Name/Initials Date 30/06/2011 Name/Initials Date 30/06/2011 Resolution Plan History
RP unique number: GI-UKEPR-CI-01-RP 0 30/06/2011 1 of 19 Approved for EDF by: A. PETIT Approved for AREVA by: C. WOOLDRIDGE Name/Initials Date 30/06/2011 Name/Initials Date 30/06/2011 Resolution Plan History
Section Qualifications of Audit teams Qualifications of Auditors Maintenance and Improvement of Competence...
 Section 9. SFI 2010-2014 Audit Procedures and Auditor Qualifications and Accreditation Updated January 2011 Section 9 Introduction... 3 1. Scope... 3 2. Normative Reference... 3 3. Terms and Definitions...
Section 9. SFI 2010-2014 Audit Procedures and Auditor Qualifications and Accreditation Updated January 2011 Section 9 Introduction... 3 1. Scope... 3 2. Normative Reference... 3 3. Terms and Definitions...
Acceptance Checklist for Abbreviated 510(k)s
s") Acceptance Checklist for Abbreviated 510(k)s (should be completed within 15 days of DCC receipt) The following information is not intended to serve as a comprehensive review. 510(k) Number: Date Received
Acceptance Checklist for Abbreviated 510(k)s (should be completed within 15 days of DCC receipt) The following information is not intended to serve as a comprehensive review. 510(k) Number: Date Received
THE GUIDE FOR ASSESSMENT OF AN EMAS ENVIRONMENTAL VERIFIER
 THE GUIDE FOR ASSESSMENT OF AN EMAS ENVIRONMENTAL VERIFIER EMAS KESKKONNATÕENDAJA HINDAMISE JUHEND EAK J18-2015 Tallinn 2015 EAK J18-2015 Page 2 of 13 Authorship and basic principles This guidance document
THE GUIDE FOR ASSESSMENT OF AN EMAS ENVIRONMENTAL VERIFIER EMAS KESKKONNATÕENDAJA HINDAMISE JUHEND EAK J18-2015 Tallinn 2015 EAK J18-2015 Page 2 of 13 Authorship and basic principles This guidance document
European Single Electronic Format (ESEF) EFRAG TEG - 10 May 2017
 EFRAG TEG - 10 May 2017") ESMA REGULAR USE ESMA32-60-171 10 May 2017 European Single Electronic Format (ESEF) EFRAG TEG - 10 May 2017 Michael Komarek Background 2 Requirements: From 1 January 2020 issuers will have to prepare their
ESMA REGULAR USE ESMA32-60-171 10 May 2017 European Single Electronic Format (ESEF) EFRAG TEG - 10 May 2017 Michael Komarek Background 2 Requirements: From 1 January 2020 issuers will have to prepare their
EU Code of Conduct on Data Centre Energy Efficiency
 EUROPEAN COMMISSION DIRECTORATE-GENERAL JRC JOINT RESEARCH CENTRE Institute for Energy Renew able and Energy Efficiency Unit EU Code of Conduct on Data Centre Energy Efficiency Introductory guide for all
EUROPEAN COMMISSION DIRECTORATE-GENERAL JRC JOINT RESEARCH CENTRE Institute for Energy Renew able and Energy Efficiency Unit EU Code of Conduct on Data Centre Energy Efficiency Introductory guide for all
20 February Accreditation of Assessment Centres
 20 February 20 Accreditation of Assessment Centres Policy Name Accreditation of Assessment Centres Document number AAC-001/ Responsible Executive Chief Director: Occupational Qualifications Quality Assurance
20 February 20 Accreditation of Assessment Centres Policy Name Accreditation of Assessment Centres Document number AAC-001/ Responsible Executive Chief Director: Occupational Qualifications Quality Assurance
Report. Certificate M6A SIMATIC Safety System
 Report to the Certificate M6A 067803 0019 Safety-Related Programmable Systems SIMATIC Safety System Manufacturer: Siemens AG Gleiwitzer Str. 555 D-90475 Nürnberg Revision 2.1 dated 2018-09-25 Testing Body:
Report to the Certificate M6A 067803 0019 Safety-Related Programmable Systems SIMATIC Safety System Manufacturer: Siemens AG Gleiwitzer Str. 555 D-90475 Nürnberg Revision 2.1 dated 2018-09-25 Testing Body:
Continuing Professional Education (CPE) CPE Rules for Pesticide Advisors
 CPE Rules for Pesticide Advisors") Irish Agricultural Supply Industry Standards CPE Rules for Pesticide Advisors IASIS Limited 2015 IASIS Ltd., 31A Ravens Rock Road, Sandyford Industrial Estate, Dublin 18 +353 (0)1 293 0021 +353 (0)1 293
Irish Agricultural Supply Industry Standards CPE Rules for Pesticide Advisors IASIS Limited 2015 IASIS Ltd., 31A Ravens Rock Road, Sandyford Industrial Estate, Dublin 18 +353 (0)1 293 0021 +353 (0)1 293
PROTERRA CERTIFICATION PROTOCOL V2.2
 PROTERRA CERTIFICATION PROTOCOL V2.2 TABLE OF CONTENTS 1. Introduction 2. Scope of this document 3. Definitions and Abbreviations 4. Approval procedure for Certification Bodies 5. Certification Requirements
PROTERRA CERTIFICATION PROTOCOL V2.2 TABLE OF CONTENTS 1. Introduction 2. Scope of this document 3. Definitions and Abbreviations 4. Approval procedure for Certification Bodies 5. Certification Requirements
Standard Development Timeline
 Standard Development Timeline This section is maintained by the drafting team during the development of the standard and will be removed when the standard is adopted by the NERC Board of Trustees (Board).
Standard Development Timeline This section is maintained by the drafting team during the development of the standard and will be removed when the standard is adopted by the NERC Board of Trustees (Board).
Notified Body perspective: Overcoming common pitfalls observed in industry when following ISO 10993
 Notified Body perspective: Overcoming common pitfalls observed in industry when following ISO 10993 Informa Life Sciences Conference Biocompatibility for Medical Devices Amsterdam, 28-29 November 2017
Notified Body perspective: Overcoming common pitfalls observed in industry when following ISO 10993 Informa Life Sciences Conference Biocompatibility for Medical Devices Amsterdam, 28-29 November 2017
ANNEXES. to the. Proposal for a Regulation of the European Parliament and of the Council
 EUROPEAN COMMISSION Brussels, 17.4.2018 COM(2018) 225 final ANNEXES 1 to 3 ANNEXES to the Proposal for a Regulation of the European Parliament and of the Council on European Production and Preservation
EUROPEAN COMMISSION Brussels, 17.4.2018 COM(2018) 225 final ANNEXES 1 to 3 ANNEXES to the Proposal for a Regulation of the European Parliament and of the Council on European Production and Preservation
List of EA Publications. Documents
 EA/INF-01 List of EA Publications and International Documents Publication Reference EA-INF/01: 2014 List of EA Publications And International Documents PURPOSE This publication gives the list of EA documents
EA/INF-01 List of EA Publications and International Documents Publication Reference EA-INF/01: 2014 List of EA Publications And International Documents PURPOSE This publication gives the list of EA documents
The European Single Electronic Format (ESEF)
") ESMA REGULAR USE 4 July 2018 The European Single Electronic Format (ESEF) TEG / CFSS meeting Objectives set out in the Transparency Directive Directive 2004/109/EC as revised by Directive 2013/50/EU (relevant
ESMA REGULAR USE 4 July 2018 The European Single Electronic Format (ESEF) TEG / CFSS meeting Objectives set out in the Transparency Directive Directive 2004/109/EC as revised by Directive 2013/50/EU (relevant
ACCREDITATION COMMISSION FOR CONFORMITY ASSESSMENT BODIES
 ACCREDITATION COMMISSION FOR CONFORMITY ASSESSMENT BODIES ACCREDITATION SCHEME MANUAL Document Title: Document Number: Various Accreditation Schemes ACCAB-ASM-7.0 CONTROLLED COPY Revision Number Revision
ACCREDITATION COMMISSION FOR CONFORMITY ASSESSMENT BODIES ACCREDITATION SCHEME MANUAL Document Title: Document Number: Various Accreditation Schemes ACCAB-ASM-7.0 CONTROLLED COPY Revision Number Revision
Certification program PCWU-3
 The certification program of utility products type 3 of the certification program according to PN-EN ISO/IEC 17067 Number: Page: 1 z 8 MS-0013527 Is valid from: 01.03.2016 Prepared: Tomasz Marcinek Approved:
The certification program of utility products type 3 of the certification program according to PN-EN ISO/IEC 17067 Number: Page: 1 z 8 MS-0013527 Is valid from: 01.03.2016 Prepared: Tomasz Marcinek Approved:
Procedure for Network and Network-related devices
 Lloyd s Register Type Approval System Type Approval Requirements for components within Cyber Enabled Systems on board Ships Procedure for Network and Network-related devices September 2017 1 Reference:
Lloyd s Register Type Approval System Type Approval Requirements for components within Cyber Enabled Systems on board Ships Procedure for Network and Network-related devices September 2017 1 Reference:
Introduction to UKAS Accreditation Fire Scene Development Programme. David Compton November 2017
 Introduction to UKAS Accreditation Fire Scene Development Programme David Compton November 2017 What is UKAS Accreditation? Procedure by which an authoritative body gives formal recognition that a body
Introduction to UKAS Accreditation Fire Scene Development Programme David Compton November 2017 What is UKAS Accreditation? Procedure by which an authoritative body gives formal recognition that a body
Accreditation Criteria For Conformity Assessment Bodies
 Page 1 of 8 Reviewed by: Getnet Tsigemalak Approved by: Araya Fesseha Position: Quality Manager Position: Director General Signature: Signature: Contents Page 1 Purpose and Scope... 2 2 References... 2
Page 1 of 8 Reviewed by: Getnet Tsigemalak Approved by: Araya Fesseha Position: Quality Manager Position: Director General Signature: Signature: Contents Page 1 Purpose and Scope... 2 2 References... 2
Balancing energy and environmental demands
 Balancing energy and environmental demands Solutions that optimise the safety and performance of conventional power plants and power station systems. TÜV SÜD South Asia Meet global energy demands As demand
Balancing energy and environmental demands Solutions that optimise the safety and performance of conventional power plants and power station systems. TÜV SÜD South Asia Meet global energy demands As demand
Data Processor Agreement
 Data Processor Agreement Data Controller: Customer located within the EU (the Data Controller ) and Data Processor: European Representative Company: ONE.COM (B-one FZ-LLC) One.com A/S Reg.no. Reg.no. 19.958
Data Processor Agreement Data Controller: Customer located within the EU (the Data Controller ) and Data Processor: European Representative Company: ONE.COM (B-one FZ-LLC) One.com A/S Reg.no. Reg.no. 19.958
European Single Electronic Format (ESEF) Eurofiling workshop 8 June 2017
 Eurofiling workshop 8 June 2017") PUBLIC ESMA32-60-177 8 June 2017 European Single Electronic Format (ESEF) Eurofiling workshop 8 June 2017 Michael Komarek Background 2 Requirements: 2013 the Transparency Directive was amended to require
PUBLIC ESMA32-60-177 8 June 2017 European Single Electronic Format (ESEF) Eurofiling workshop 8 June 2017 Michael Komarek Background 2 Requirements: 2013 the Transparency Directive was amended to require
IQ Level 4 Award in Understanding the External Quality Assurance of Assessment Processes and Practice (QCF) Specification
 Specification") IQ Level 4 Award in Understanding the External Quality Assurance of Assessment Processes and Practice (QCF) Specification Regulation No: 600/5528/5 Page 1 of 15 Contents Page Industry Qualifications...
IQ Level 4 Award in Understanding the External Quality Assurance of Assessment Processes and Practice (QCF) Specification Regulation No: 600/5528/5 Page 1 of 15 Contents Page Industry Qualifications...