MS-FINDER tutorial. Last edited in Aug 10, 2018
|
|
|
- Logan Carter
- 5 years ago
- Views:
Transcription
 and liquid chromatography coupled with electrospray ionization- (ESI-) tandem mass spectrometer (LC/MS/MS) are the")
1 MS-FINDER tutorial Last edited in Aug 10, 2018 Abstract The purpose of metabolomics is to perform the comprehensive analysis for small biomolecules of living organisms. Gas chromatography coupled with electron ionization mass spectrometer (GC/MS) and liquid chromatography coupled with electrospray ionization- (ESI-) tandem mass spectrometer (LC/MS/MS) are the preferred tools for untargeted metabolomics. Currently, the main bottleneck of GC/MS- and LC/MS/MS based untargeted analysis is compound identification due to the limitation of EI-MS and MS/MS records of authentic standard. MS-FINDER was launched as a universal program for compound annotation that supports EI-MS (GC/MS) and MS/MS spectral mining. First, MS-FINDER aims to provide solutions for 1) formula predictions, 2) fragment annotations, and 3) structure elucidations by means of unknown spectra. In addition, the program can annotate your unknowns by the public spectral databases such as MassBank, LipidBlast, and GNPS. MS-FINDER has been developed as the collaborative work between Prof. Masanori Arita team (RIKEN, Reifycs Inc.) and Prof. Oliver Fiehn team (UC Davis) supported by the JST/NSF SICORP Metabolomics for the low carbon society project. Hiroshi Tsugawa RIKEN Center for Sustainable Resource Science hiroshi.tsugawa@riken.jp MS-FINDER screenshot 1
2 Table of contents Software environments... 3 Required programs... 4 Acceptable ASCII formats... 5 MSP format for MS/MS... 6 MSP format for EI-MS... 7 MAT format... 8 Adduct ion format: [M+Na]+, [M+2H]2+, [M-2H2O+H]+, [2M+FA-H]-, etc User defined structure database format Specific field to fix the formula element count Import queries A. From a folder which includes MSP or MAT format files B. From the graphical user interface of the MS-FINDER program C. From the MS-DIAL program Parameter setting Method tab Mass spectrum tab Formula finder tab Structure finder tab Data source tab Compound annotation by in silico fragmenter Compound annotation by searching spectral databases Compound annotation (batch analysis) Peak assignment (single) Peak assignment (batch job) Mouse function Export Help
3 Software environments Windows OS (.NET Framework 4.5 or later): Windows 7 or later RAM: 8.0 GB or more 3
4 Required programs MS-FINDER Download link: MS-FINDER can be used as the local software program in Windows PC. The program can import ASCII format files including MSP (EI-MS and MS/MS) or improved MSP (both MS and MS/MS, the file extension must be MAT.). In addition, the users can directly make the query in the MS-FINDER graphical user interface. Moreover, this program can be called from the MS- DIAL program which is downloadable at DIAL/index.html. 4
5 Acceptable ASCII formats This program accepts two file extensions, i.e. MSP or MAT formatted by the following explanations. Unknown queries should be separately stored in the ASCII file: the MSP or MAP file CANNOT store multi compound records in the single file. The format of MSP basically follows the NIST MS search manual. Link: 5
 Mass spectrum is supposed to be imported as MS/MS. If you want to perform the MS/MS peak annotation with the known structure, prepare two fields including FORMULA and SMILES.")
6 MSP format for MS/MS Required fields NAME: PRECURSORMZ: PRECURSORTYPE: IONMODE: (Positive or Negative) Num Peaks: m/z intensity pair (tab, comma, space can be used as the delimiter.) Mass spectrum is supposed to be imported as MS/MS. If you want to perform the MS/MS peak annotation with the known structure, prepare two fields including FORMULA and SMILES. Please note that the formula and SMILES of the neutralized structure should be prepared. MSP example 6
7 MSP format for EI-MS Required fields NAME: IONMODE: (Positive or Negative) Num Peaks: m/z intensity pair (tab, comma, space can be used as the delimiter.) The fields are the minimum requirement for searching spectral databases. In the case that you want to perform formula predictions and structure elucidations in EI-MS data, two files PRECURSORMZ: and PRECURSORTYPE: must be required. 7
8 MAT format The MAT format was defined as the improved version of MSP in the MS-FIDNER program to store both MS1 and MS/MS spectra in the same file. The survey scan MS data should be required to calculate isotopic ion score for formula predictions. Importantly, for EI-MS spectra, put your spectra into both MS1- and MS2 fields for the calculation of isotopic ratio and fragment ion similarities, respectively. Required fields NAME: PRECURSORMZ: PRECURSORTYPE: IONMODE: MSTYPE: Num Peaks: m/z intensity pair (tab, comma, space can be used as the delimiter.) Three fields including MSTYPE, Num Peaks, and m/z intensity pair should be SERIALLY stored. If you type MSTYPE: MS1, the spectrum written from next field should be recognized as the survey scan MS (MS1). If you type MSTYPE: MS2, next spectrum should be recognized as the MS/MS spectrum. EI-MS spectra must be stored in both MS1 and MS2, which is the requirement of MS-FINDER. Both field (MSTYPE: MS1 and MSTYPE: MS2) is not necessary for this program, i.e. the users can import the ASCII file as only MS1 spectrum or as only MS/MS spectrum record. Users may prepare the MAT or MSP files without any spectrum record. In such case, the formula prediction will be performed by means of mass accuracy and database criteria. If you want to perform the MS/MS peak annotation with the known structure, prepare two fields including FORMULA and SMILES. The formula and SMILES of the neutralized structure should be made. 8
9 MAT example 9
10 Adduct ion format: [M+Na]+, [M+2H]2+, [M-2H2O+H]+, [2M+FA-H]-, etc. 1. The parentheses [ and ] must be used to bracket the ion information. 2. The char + and - must be required after ']' and the number must be written before + or When you want to define the organic formula like C6H12O5, you have to write it without any replicate elements or parentheses. For example, the descriptions like [M+C2H5COOH-H]- or [M+H+(CH3)3SiOH]+ are not accepted. 4. The beginning figure of organic formula like '2'H2O is recognized as the H2O 2. Again, never use 2(H2O) for that. 5. Sequential equations are acceptable: [2M+H-C6H12O5+Na]2+ (very apt.) 6. Radical ion can be described by.(dot) after + or like [M]+. And [M-CH3]+. as adduct format. 7. MS-FINDER accepts some abbreviations or common organic formulas for adduct types as follows. For Acetonitrile: ACN, CH3CN For Methanol: CH3OH For Isopropanol: IsoProp, C3H7OH For Dimethyl sulfoxide: DMSO For Formic acid: FA, HCOOH For Acetic acid: Hac, CH3COOH For Trifluoroacetic acid: TFA, CFCOOH 10

11 User defined structure database format MS-FINDER supports the structure elucidations from the candidates that users provide. The following format file should be prepared as tab-delimited text file. The identifiers of InChIKey, short InChIKey, and database ID are not required, but the values must be filled by some mimic values. The files of exact mass, formula, and SMILES must be prepared. 11


12 Specific field to fix the formula element count MS-FINDER recently accepts the filtering field for molecular formula prediction. For example, if you can perform fully labeled stable isotope experiment, the element count for CHNOS can be determined by checking the mass shift between non-labeled and labeled sample data. The fields described in the below figure can be written in MSP and MAT files. 12


13 Import queries There are two ways to import unknown queries. A. From a folder which includes MSP or MAT format files 1. File -> import 2. Select a folder containing MSP or MAT files. 13


14 B. From the graphical user interface of the MS-FINDER program 1. File -> Create a query 2. Fill in the form to make a query. Required files Folder path File name Precursor m/z 14


15 C. From the MS-DIAL program The MS-DIAL program which has been reported as software for data processing of LC/MS/MS can call the MS-FINDER program directly. On the first time when you call the MS-FINDER program at MS-DIAL, please select the file path of MS-FINDER via GUI. 15

16 Parameter setting Method tab MS-FINDER provides two options for compound annotation: one is by spectral databases, and the other is by formula- and structure finder programs using in silico fragmenter. You can simultaneously check both spectral database search and formula prediction and structure elucidation by in silico fragmenter options, and the result of spectral database search has priority for ranking structures. The internal experimental library is stored in EIMS-DBs-vs*.egm and MSMS-DBs-vs*.etm of Resources folder as NIST MSP format. If Formula finder > TMS-MeOX derivative compound is checked, EIMS database will be used; otherwise, MSMS database is used. The in silico library for lipids (LipidBlast) is stored in MSDIAL-LipidDBs-vs*.lbm of Resources folder. Select the appropriate solvent condition for searching your unknowns. The user-defined spectral database must be formatted by NIST MSP. If Precursor oriented spectral search is checked, the structure candidates will be filtered out by the precursor m/z of spectral records in combination with MS1 tolerance value; otherwise, all of spectral records will be used. Uncheck this option if you want to search EI spectral databases. 16
17 Mass spectrum tab Mass tolerance (MS1): the mass tolerance to generate formula candidates. Mass tolerance (MS/MS): the mass tolerance for matching experimental- and reference fragments. Relative abundance cut off: The product ions more than this parameter on the basis of base peak ion are utilized for the product ion matching. *For EI-MS spectra, set the same tolerance into MS1 and MS2. 17
18 Formula finder tab Formula calculation setting: You can set the parameters for formula calculation. LOWIS and SENIOR check: to generate formula candidates that match the valence rules of formula elements. The valences of hetero atoms, i.e. N, O, S, and P are currently set to 3, 2, 6, and 5, respectively. Isotopic ratio tolerance: to calculate the isotopic score. The tolerance should be utilized as the sigma value for the Gaussian scoring as described in the MS-FINDER paper. Element ratio check: to generate formula candidates that satisfy every element ratios (ex. H/C ratio should be between 0 and 3.33 for Common range (99.7%) restriction. ) as described in the MS- FINDER paper. Element probability check: to generate formula candidates that satisfy the heuristic rules as described in the Seven Golden Rules paper. For example, if a formula candidate contains the following element counts, i.e. NOPS all > 1, the element counts of N, O, P, and S should be less than 9, 19, 3, and 2, respectively. Element selection: to generate formula candidates that just contain the elements selected by the users. Check TMS-MEOX derivative compound if you want to annotate EI-MS spectra. Result cut off: formula candidates ranked by the MS-FINDER program will be reported within up to this number. 18
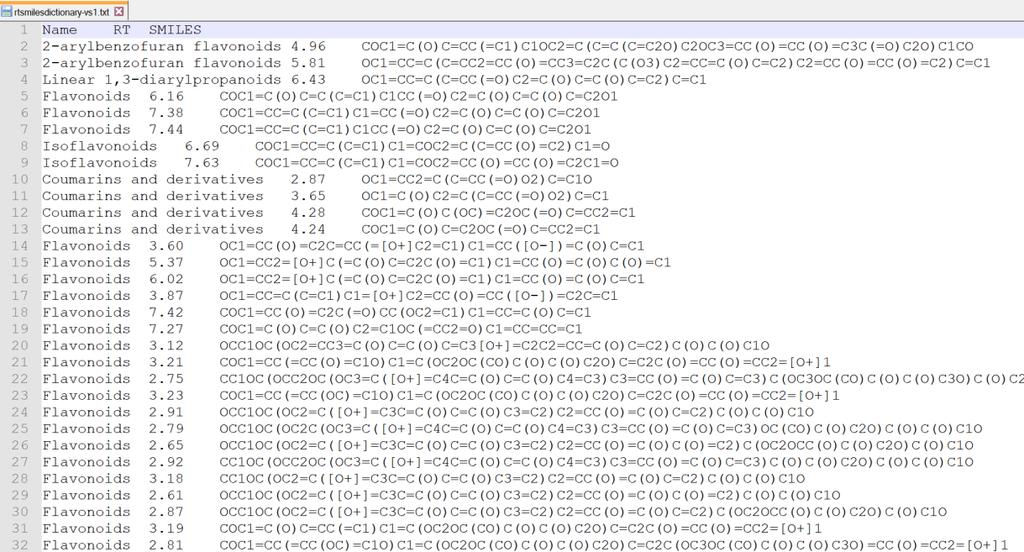
19 Structure finder tab Here is the parameter setting for in silico fragmenter. Tree depth: the limitation of in silico cleavages, i.e. if the user sets 2, the MS-FINDER program generates the fragments until product ions of a product ion. The current MS-FINDER program can utilize the fragment ion library for EI-MS spectral mining which is stored in *.eif (recommended to use). Result cut off: structure candidates ranked by the MS-FINDER program will be reported within up to this number. XLogP based RT prediction and cut off setting for structure elucidation Recently, MS-FINDER provides a simple RT prediction function using XLogP calculated by CDK. If you prepare the tab-delimited text format file containing (first column) metabolite name, (second column) retention time (min), and (third column) SMILES code as described below, the predicted retention time is calculated for searching structure candidates. Retention time setting for spectral searching This function is for spectral searching. If your MSP or MAT files contain the retention time or retention index information, you can use the RT or RI filtering by means of this checkbox. 19
20 20
21 Data source tab Local databases: currently, total 14 metabolome databases are prepared in the MS-FINDER program which is stored in *.esd. The local databases selected by users will be used to retrieve the structure data. Please see user defined database format section for searching your own structure candidates. MINEs (Metabolic In silico Network Expansions) and PubChem online settings: If the user selects Never use it, the structure candidates will be picked up just from the local databases. If only use when there is no query in the below DBs is selected, the structure data of MINE and PubChem compound databases will be retrieved when no structures can be found in the local databases. If always use it. is selected, the MS-FINDER program always retrieve the structure data from MINE and PubChem databases in addition to local databases. 21

 The formula candidate checked in Select column is supposed to be examined at structure finder program. B) The isotopic ions will be displayed by I button at the upper mass spectrum window.")
22 Compound annotation by in silico fragmenter The general workflow of MS-FINDER is described here. (Batch analysis is shown below) 1. The formula prediction is executed by double click at the title name of File navigator. The detail of formula calculations is described in our paper. *The point of formula prediction is to correctly select the precursor type (adduct ion) and to set parameters for picking up the correct formulas. A) The formula candidate checked in Select column is supposed to be examined at structure finder program. B) The isotopic ions will be displayed by I button at the upper mass spectrum window. C) The result of formula assignment in product ions will be displayed by P button at the bottom spectrum window. D) The annotation result of neutral losses will be displayed by N button at bottom window. 22
 The formula candidate checked in Select column is supposed to be examined at structure finder program. B) Total score of structure finder is the total of formula- and structure scores.")
23 2. The structure finder will be executed by right click at the formula result table followed by clicking Search the structure. A) The formula candidate checked in Select column is supposed to be examined at structure finder program. B) Total score of structure finder is the total of formula- and structure scores. Therefore, even if a formula score is greater than the others, the other structure from another formula candidate may become the top candidate in the structure finder program. C) The MS-FINDER program integrates the structures having the same molecular skeleton by its InChIKey (first 14 characters). Its representative structure will be determined by the number of synonymous in PubChem repository. 23

24 Compound annotation by searching spectral databases It s very simple. Double click the unknown record that you want to annotate. The below is the examples for searching EI-MS spectral database and LipidBlast database. 24
 A) If you want to perform the batch job for both formula predictions and structure elucidations, please check Both processes.")
25 Compound annotation (batch analysis) 1. Analysis -> Compound annotation (batch job) A) If you want to perform the batch job for both formula predictions and structure elucidations, please check Both processes. Also, add the number in Top N hits textbox where the formula candidates generated by the formula finder program are supposed to be searched. B) If you want to perform the batch job for formula predictions, please just check molecular formula finder. C) If you want to perform the structure finder program, please just check structure finder. Here, the formula candidates checked by the users are supposed to be examined. Also, if the formula finder is not executed before this analysis, the queries will be passed. 25


26 Peak assignment (single) The MS-FINDER program can be used as the peak assignment tool to assign substructures in the MS/MS spectrum from user-defined structure. 1. Analysis -> Peak assignment (single) 2. Select the query file that you want to analyze. 3. Add both formula and SMILES into the textboxes as the neutralized form. 4. The result will be generated as shown below. 26

27 Peak assignment (batch job) Analysis-> Peak assignment (batch job) To use this program, please make sure that the record in MSP or MAT files should contains the respective FORMULA and SMILES fields. Otherwise, the program will be passed for records not having their fields. The SMILES or FORMULA records can be added in File information textboxes of MS-FINDER GUI. 27
 Mouse wheel: zoom in and out E) Right click: popup context menu")
28 Mouse function A) Mouse right click (or hold) and move: zoom in and out B) Mouse left click (or hold) and move: select and scroll C) Mouse left double click: reset range and select files in the file navigator D) Mouse wheel: zoom in and out E) Right click: popup context menu 28

29 Export The result of formula and structure finders can be exported from this option. Currently, the top 10 candidates will be automatically exported. However, you can also check the details of result as the ASCII file. It means that the MS-FINDER program is supposed to generate the FGT file containing formula results in the same directory as the project folder. Also, the program generates the respective folder containing the SFD file (per formula candidate) which stores the result of structure finder program. 29

30 Help You can check the version of MS-FINDER. 30
MS-FINDER tutorial. Last edited in Sep. 10, 2018
 MS-FINDER tutorial Last edited in Sep. 10, 2018 Abstract The purpose of metabolomics is to perform the comprehensive analysis for small biomolecules of living organisms. Gas chromatography coupled with
MS-FINDER tutorial Last edited in Sep. 10, 2018 Abstract The purpose of metabolomics is to perform the comprehensive analysis for small biomolecules of living organisms. Gas chromatography coupled with
MRMPROBS tutorial. Hiroshi Tsugawa RIKEN Center for Sustainable Resource Science
 MRMPROBS tutorial Edited in 2014/10/6 Introduction MRMPROBS is a tool for the analysis of data from multiple reaction monitoring (MRM)- or selected reaction monitoring (SRM)-based metabolomics studies.
MRMPROBS tutorial Edited in 2014/10/6 Introduction MRMPROBS is a tool for the analysis of data from multiple reaction monitoring (MRM)- or selected reaction monitoring (SRM)-based metabolomics studies.
MRMPROBS tutorial. Hiroshi Tsugawa RIKEN Center for Sustainable Resource Science MRMPROBS screenshot
 MRMPROBS tutorial Edited in 2016/11/16 Introduction MRMPROBS is launched as a universal program for targeted metabolomics using not only multiple reaction monitoring (MRM)- or selected reaction monitoring
MRMPROBS tutorial Edited in 2016/11/16 Introduction MRMPROBS is launched as a universal program for targeted metabolomics using not only multiple reaction monitoring (MRM)- or selected reaction monitoring
Agilent G6854 MassHunter Personal Pesticide Database
 Agilent G6854 MassHunter Personal Pesticide Database Quick Start Guide What is MassHunter Personal Pesticide Database? 2 Installation 3 Main Window 4 Getting Started 11 Database operations 12 Searching
Agilent G6854 MassHunter Personal Pesticide Database Quick Start Guide What is MassHunter Personal Pesticide Database? 2 Installation 3 Main Window 4 Getting Started 11 Database operations 12 Searching
MassHunter Personal Compound Database and Library Manager for Forensic Toxicology
 MassHunter Personal Compound Database and Library Manager for Forensic Toxicology Quick Start Guide What is MassHunter Personal Compound Database and Library Manager? 2 Installation 3 Main Window 4 Getting
MassHunter Personal Compound Database and Library Manager for Forensic Toxicology Quick Start Guide What is MassHunter Personal Compound Database and Library Manager? 2 Installation 3 Main Window 4 Getting
Pathway Analysis of Untargeted Metabolomics Data using the MS Peaks to Pathways Module
 Pathway Analysis of Untargeted Metabolomics Data using the MS Peaks to Pathways Module By: Jasmine Chong, Jeff Xia Date: 14/02/2018 The aim of this tutorial is to demonstrate how the MS Peaks to Pathways
Pathway Analysis of Untargeted Metabolomics Data using the MS Peaks to Pathways Module By: Jasmine Chong, Jeff Xia Date: 14/02/2018 The aim of this tutorial is to demonstrate how the MS Peaks to Pathways
Skyline High Resolution Metabolomics (Draft)
") Skyline High Resolution Metabolomics (Draft) The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally
Skyline High Resolution Metabolomics (Draft) The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally
Agilent G6825AA METLIN Personal Metabolite Database for MassHunter Workstation
 Agilent G6825AA METLIN Personal Metabolite Database for MassHunter Workstation Quick Start Guide This guide describes how to install and use METLIN Personal Metabolite Database for MassHunter Workstation.
Agilent G6825AA METLIN Personal Metabolite Database for MassHunter Workstation Quick Start Guide This guide describes how to install and use METLIN Personal Metabolite Database for MassHunter Workstation.
Skyline Targeted MS/MS
 Skyline Targeted MS/MS Skyline now supports several methods of extracting chromatography-based quantitative measurements from the raw data files of full-scan mass spectrometers, such as ion trap and Q-TOF
Skyline Targeted MS/MS Skyline now supports several methods of extracting chromatography-based quantitative measurements from the raw data files of full-scan mass spectrometers, such as ion trap and Q-TOF
 HOW TO PERFORM LC-MS ANNOTATIONS? W4M Core Team 1 «LC-MS ANNOTATION» MODULES IN W4M The «LCMS Annotation» modules allow you to perform: «Adducts» annotation «Chemical» annotation: Meta-engine (Chemspider)
HOW TO PERFORM LC-MS ANNOTATIONS? W4M Core Team 1 «LC-MS ANNOTATION» MODULES IN W4M The «LCMS Annotation» modules allow you to perform: «Adducts» annotation «Chemical» annotation: Meta-engine (Chemspider)
Tutorial 7: Automated Peak Picking in Skyline
 Tutorial 7: Automated Peak Picking in Skyline Skyline now supports the ability to create custom advanced peak picking and scoring models for both selected reaction monitoring (SRM) and data-independent
Tutorial 7: Automated Peak Picking in Skyline Skyline now supports the ability to create custom advanced peak picking and scoring models for both selected reaction monitoring (SRM) and data-independent
TraceFinder Analysis Quick Reference Guide
 TraceFinder Analysis Quick Reference Guide This quick reference guide describes the Analysis mode tasks assigned to the Technician role in the Thermo TraceFinder 3.0 analytical software. For detailed descriptions
TraceFinder Analysis Quick Reference Guide This quick reference guide describes the Analysis mode tasks assigned to the Technician role in the Thermo TraceFinder 3.0 analytical software. For detailed descriptions
Agilent MassHunter Qualitative Data Analysis
 Agilent MassHunter Qualitative Data Analysis Qualitative Navigator B.08.00 Presenters: Kevin Costalunga Stephen Harnos With Matt Leyden & Kevin Costalunga 1 MassHunter Webinar Series MassHunter Qualitative
Agilent MassHunter Qualitative Data Analysis Qualitative Navigator B.08.00 Presenters: Kevin Costalunga Stephen Harnos With Matt Leyden & Kevin Costalunga 1 MassHunter Webinar Series MassHunter Qualitative
Agilent G6854AA MassHunter Personal Compound Database
 Agilent G6854AA MassHunter Personal Compound Database Quick Start Guide This guide describes how to install and use MassHunter Personal Compound Database. Where to find more information What is Personal
Agilent G6854AA MassHunter Personal Compound Database Quick Start Guide This guide describes how to install and use MassHunter Personal Compound Database. Where to find more information What is Personal
Protein Deconvolution Quick Start Guide
 Protein Deconvolution Quick Start Guide The electrospray ionization (ESI) of intact peptides and proteins produces mass spectra containing a series of multiply charged ions with associated mass-to-charge
Protein Deconvolution Quick Start Guide The electrospray ionization (ESI) of intact peptides and proteins produces mass spectra containing a series of multiply charged ions with associated mass-to-charge
RMassBank: Run-through the Principles and Workflow in R
 RMassBank: Run-through the Principles and Workflow in R Emma Schymanski Michael Stravs, Heinz Singer & Juliane Hollender Eawag, Dübendorf, Switzerland Steffen Neumann, Erik Müller: IPB Halle, Germany Tobias
RMassBank: Run-through the Principles and Workflow in R Emma Schymanski Michael Stravs, Heinz Singer & Juliane Hollender Eawag, Dübendorf, Switzerland Steffen Neumann, Erik Müller: IPB Halle, Germany Tobias
MSFragger Manual. (build )
") MSFragger Manual (build 20170103.0) Introduction MSFragger is an ultrafast database search tool for peptide identifications in mass spectrometry-based proteomics. It differs from conventional search engines
MSFragger Manual (build 20170103.0) Introduction MSFragger is an ultrafast database search tool for peptide identifications in mass spectrometry-based proteomics. It differs from conventional search engines
LC-MS Data Pre-Processing. Xiuxia Du, Ph.D. Department of Bioinformatics and Genomics University of North Carolina at Charlotte
 LC-MS Data Pre-Processing Xiuxia Du, Ph.D. Department of Bioinformatics and Genomics University of North Carolina at Charlotte Outline Raw LC-MS data - Profile and centroid data - Mass vs. retention time
LC-MS Data Pre-Processing Xiuxia Du, Ph.D. Department of Bioinformatics and Genomics University of North Carolina at Charlotte Outline Raw LC-MS data - Profile and centroid data - Mass vs. retention time
Data Processing for Small Molecules
 Data Processing for Small Molecules Basic Metabolomics Workflows Metabolomics: the apogee of the omics trilogy Gary J. Patti, Oscar Yanes and Gary Siuzdak Molecular Cell Biology, 2012, 13, 263-269 MATTHEW
Data Processing for Small Molecules Basic Metabolomics Workflows Metabolomics: the apogee of the omics trilogy Gary J. Patti, Oscar Yanes and Gary Siuzdak Molecular Cell Biology, 2012, 13, 263-269 MATTHEW
Tutorial 2: Analysis of DIA/SWATH data in Skyline
 Tutorial 2: Analysis of DIA/SWATH data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantification.
Tutorial 2: Analysis of DIA/SWATH data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantification.
De Novo Peptide Identification
 De Novo Peptide Identification When a suitable sequence database is not available for your sample of interest, identification of your acquired fragmentation mass spectra becomes quite difficult. Indeed,
De Novo Peptide Identification When a suitable sequence database is not available for your sample of interest, identification of your acquired fragmentation mass spectra becomes quite difficult. Indeed,
Progenesis CoMet User Guide
 Progenesis CoMet User Guide Analysis workflow guidelines for version 2.0 Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using CoMet?... 3 LC-MS Data used in this
Progenesis CoMet User Guide Analysis workflow guidelines for version 2.0 Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using CoMet?... 3 LC-MS Data used in this
Building Agilent GC/MSD Deconvolution Reporting Libraries for Any Application Technical Overview
 Building Agilent GC/MSD Deconvolution Reporting Libraries for Any Application Technical Overview Authors Xiaofei Ping Agilent Technologies (Shanghai) Co Ltd 412 Yinglun Road, Shanghai, 200131 P.R. China
Building Agilent GC/MSD Deconvolution Reporting Libraries for Any Application Technical Overview Authors Xiaofei Ping Agilent Technologies (Shanghai) Co Ltd 412 Yinglun Road, Shanghai, 200131 P.R. China
MassBank for NORMAN. Achievements in 2011 and further proposed steps for identification of unknowns
 MassBank for NRMAN Achievements in 2011 and further proposed steps for identification of unknowns Tobias Schulze NRMAN GA 2011, Stockholm 22-23 November 2011 tobias.schulze@ufz.de Background and objective
MassBank for NRMAN Achievements in 2011 and further proposed steps for identification of unknowns Tobias Schulze NRMAN GA 2011, Stockholm 22-23 November 2011 tobias.schulze@ufz.de Background and objective
Discussion on harmonisation potential and needs. Peter Haglund Umeå University, Sweden
 Discussion on harmonisation potential and needs Peter Haglund Umeå University, Sweden Why harmonize? Avoid loss of information Comparability of data Exchange of data Data mining Emerging (hazardous) pollutant
Discussion on harmonisation potential and needs Peter Haglund Umeå University, Sweden Why harmonize? Avoid loss of information Comparability of data Exchange of data Data mining Emerging (hazardous) pollutant
Application Note. Abstract. Authors. Forensic Toxicology
 An Application Kit for the Screening of Samples for Analytes of Forensic Toxicological Interest using TOF or Q-TOF LC/MS with a Personal Forensic Toxicology Database Application Note Forensic Toxicology
An Application Kit for the Screening of Samples for Analytes of Forensic Toxicological Interest using TOF or Q-TOF LC/MS with a Personal Forensic Toxicology Database Application Note Forensic Toxicology
Agilent MassHunter Workstation Software 7200 Accurate-Mass Quadrupole Time of Flight GC/MS
 Agilent MassHunter Workstation Software 7200 Accurate-Mass Quadrupole Time of Flight GC/MS Familiarization Guide Before you begin 3 Prepare your system 3 Prepare the samples required for data acquisition
Agilent MassHunter Workstation Software 7200 Accurate-Mass Quadrupole Time of Flight GC/MS Familiarization Guide Before you begin 3 Prepare your system 3 Prepare the samples required for data acquisition
Curatr: a web application for creating, curating, and sharing a mass spectral library
 Curatr: a web application for creating, curating, and sharing a mass spectral library Andrew Palmer (1), Prasad Phapale (1), Dominik Fay (1), Theodore Alexandrov (1,2) (1) European Molecular Biology Laboratory,
Curatr: a web application for creating, curating, and sharing a mass spectral library Andrew Palmer (1), Prasad Phapale (1), Dominik Fay (1), Theodore Alexandrov (1,2) (1) European Molecular Biology Laboratory,
Annotation of LC-MS metabolomics datasets by the metams package
 Annotation of LC-MS metabolomics datasets by the metams package Pietro Franceschi April 30, 2018 1 Introduction metams is designed to perform the analysis of LC-MS and GC-MSbased metabolomics assays. For
Annotation of LC-MS metabolomics datasets by the metams package Pietro Franceschi April 30, 2018 1 Introduction metams is designed to perform the analysis of LC-MS and GC-MSbased metabolomics assays. For
NIST MS AUTOIMP feature looks for and finds secondary locator file.
 INTRODUCTION This document describes how to start the NIST MS (Mass Spectral) Search Program Version 2.0 and higher or the AMDIS (Automated Mass spectral Deconvolution and Identification) program from
INTRODUCTION This document describes how to start the NIST MS (Mass Spectral) Search Program Version 2.0 and higher or the AMDIS (Automated Mass spectral Deconvolution and Identification) program from
Agilent G2721AA Spectrum Mill MS Proteomics Workbench Quick Start Guide
 Agilent G2721AA Spectrum Mill MS Proteomics Workbench Quick Start Guide A guide to the Spectrum Mill workbench Use this reference for your first steps with the Spectrum Mill workbench. What is the Spectrum
Agilent G2721AA Spectrum Mill MS Proteomics Workbench Quick Start Guide A guide to the Spectrum Mill workbench Use this reference for your first steps with the Spectrum Mill workbench. What is the Spectrum
MassHunter Pesticides PCD or PCDL Quick Start Guide
 MassHunter Pesticides PCD or PCDL Quick Start Guide What is the MassHunter Pesticides PCD or PCDL? 3 Kit Contents 4 Where to find more information Before You Begin 7 Installation 7 Required reagents and
MassHunter Pesticides PCD or PCDL Quick Start Guide What is the MassHunter Pesticides PCD or PCDL? 3 Kit Contents 4 Where to find more information Before You Begin 7 Installation 7 Required reagents and
SIMAT: GC-SIM-MS Analayis Tool
 SIMAT: GC-SIM-MS Analayis Tool Mo R. Nezami Ranjbar September 10, 2018 1 Selected Ion Monitoring Gas chromatography coupled with mass spectrometry (GC-MS) is one of the promising technologies for qualitative
SIMAT: GC-SIM-MS Analayis Tool Mo R. Nezami Ranjbar September 10, 2018 1 Selected Ion Monitoring Gas chromatography coupled with mass spectrometry (GC-MS) is one of the promising technologies for qualitative
Agilent MassHunter Metabolite ID Software. Installation and Getting Started Guide
 Agilent MassHunter Metabolite ID Software Installation and Getting Started Guide Notices Agilent Technologies, Inc. 2011 No part of this manual may be reproduced in any form or by any means (including
Agilent MassHunter Metabolite ID Software Installation and Getting Started Guide Notices Agilent Technologies, Inc. 2011 No part of this manual may be reproduced in any form or by any means (including
Statistical Process Control in Proteomics SProCoP
 Statistical Process Control in Proteomics SProCoP This tutorial will guide you through the installation of SProCoP and using it to perform statistical analysis on a sample Skyline file. Getting Started
Statistical Process Control in Proteomics SProCoP This tutorial will guide you through the installation of SProCoP and using it to perform statistical analysis on a sample Skyline file. Getting Started
Panorama Sharing Skyline Documents
 Panorama Sharing Skyline Documents Panorama is a freely available, open-source web server database application for targeted proteomics assays that integrates into a Skyline proteomics workflow. It has
Panorama Sharing Skyline Documents Panorama is a freely available, open-source web server database application for targeted proteomics assays that integrates into a Skyline proteomics workflow. It has
MALDI Software Users Guide (simplified version)
") MALDI Software Users Guide (simplified version) Central Analytical Lab Ekeley E266 The Acquisition program is called Voyager Control Panel. Once you shoot your sample and determine it looks good, you transfer
MALDI Software Users Guide (simplified version) Central Analytical Lab Ekeley E266 The Acquisition program is called Voyager Control Panel. Once you shoot your sample and determine it looks good, you transfer
Package Metab. September 18, 2018
 Version 1.14.0 Date 2013-10-11 Package Metab September 18, 2018 Title Metab: An R Package for a High-Throughput Analysis of Metabolomics Data Generated by GC-MS. Author Raphael Aggio
Version 1.14.0 Date 2013-10-11 Package Metab September 18, 2018 Title Metab: An R Package for a High-Throughput Analysis of Metabolomics Data Generated by GC-MS. Author Raphael Aggio
NORMAN MassBank and beyond Status and future of activities regarding mass spectral databases
 NORMAN MassBank and beyond Status and future of activities regarding mass spectral databases NORMAN Annual General Assembly Meeting Vienna, 30 November - 01 December 2016 commons.wikimedia.org Tobias Schulze,
NORMAN MassBank and beyond Status and future of activities regarding mass spectral databases NORMAN Annual General Assembly Meeting Vienna, 30 November - 01 December 2016 commons.wikimedia.org Tobias Schulze,
An R package to process LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit)
") An R package to process LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit) Francesc Fernández-Albert, Rafael Llorach, Cristina Andrés-Lacueva, Alexandre Perera June 13, 2018 1 Abstract
An R package to process LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit) Francesc Fernández-Albert, Rafael Llorach, Cristina Andrés-Lacueva, Alexandre Perera June 13, 2018 1 Abstract
Xcalibur Library Browser
 Thermo Xcalibur Library Browser User Guide Creating and Searching Spectral Libraries Software Version 3.0 XCALI-97552 Revision A June 2013 2013 Thermo Fisher Scientific Inc. All rights reserved. Xcalibur
Thermo Xcalibur Library Browser User Guide Creating and Searching Spectral Libraries Software Version 3.0 XCALI-97552 Revision A June 2013 2013 Thermo Fisher Scientific Inc. All rights reserved. Xcalibur
Skyline MS1 Full Scan Filtering
 Skyline MS1 Full Scan Filtering The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline project. These displays allow
Skyline MS1 Full Scan Filtering The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline project. These displays allow
Skyline Targeted Method Editing
 Skyline Targeted Method Editing This tutorial will cover many of the features available in the Skyline Targeted Proteomics Environment for creating new instrument methods for Selected Reaction Monitoring
Skyline Targeted Method Editing This tutorial will cover many of the features available in the Skyline Targeted Proteomics Environment for creating new instrument methods for Selected Reaction Monitoring
Progenesis CoMet User Guide
 Progenesis CoMet User Guide Analysis workflow guidelines for version 1.0 Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using CoMet?... 3 LC-MS Data used in this
Progenesis CoMet User Guide Analysis workflow guidelines for version 1.0 Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using CoMet?... 3 LC-MS Data used in this
Skyline Targeted Method Refinement
 Skyline Targeted Method Refinement This tutorial will introduce the features available in the Skyline Targeted Proteomics Environment for refining instrument methods for Selected Reaction Monitoring (SRM,
Skyline Targeted Method Refinement This tutorial will introduce the features available in the Skyline Targeted Proteomics Environment for refining instrument methods for Selected Reaction Monitoring (SRM,
Agilent Triple Quadrupole LC/MS Peptide Quantitation with Skyline
 Agilent Triple Quadrupole LC/MS Peptide Quantitation with Skyline Workflow Guide A Peptide Optimization B Review Results in Skyline Create QQQ method in Skyline Edit Skyline settings Import peptides into
Agilent Triple Quadrupole LC/MS Peptide Quantitation with Skyline Workflow Guide A Peptide Optimization B Review Results in Skyline Create QQQ method in Skyline Edit Skyline settings Import peptides into
Skyline irt Retention Time Prediction
 Skyline irt Retention Time Prediction Predicting peptide retention time has long been of interest in targeted proteomics. As early as version 0.2, Skyline integrated the SSRCalc hydrophobicity calculator
Skyline irt Retention Time Prediction Predicting peptide retention time has long been of interest in targeted proteomics. As early as version 0.2, Skyline integrated the SSRCalc hydrophobicity calculator
OpenLynx User's Guide
 OpenLynx User s Guide OpenLynx User's Guide Version 4.0 Waters part No 715000405 Micromass Part No - 6666670 5 February, 2002 i OpenLynx User s Guide OpenLynx User s Guide The software described in this
OpenLynx User s Guide OpenLynx User's Guide Version 4.0 Waters part No 715000405 Micromass Part No - 6666670 5 February, 2002 i OpenLynx User s Guide OpenLynx User s Guide The software described in this
Corra v2.0 User s Guide
 Corra v2.0 User s Guide Corra is an open source software Licensed under the Apache License, Version 2.0 and it s source code, demo data and this guide can be downloaded at the http://tools.proteomecenter.org/corra/corra.html.
Corra v2.0 User s Guide Corra is an open source software Licensed under the Apache License, Version 2.0 and it s source code, demo data and this guide can be downloaded at the http://tools.proteomecenter.org/corra/corra.html.
Skyline Targeted Method Refinement
 Skyline Targeted Method Refinement This tutorial will introduce the features available in the Skyline Targeted Proteomics Environment for refining instrument methods for Selected Reaction Monitoring (SRM,
Skyline Targeted Method Refinement This tutorial will introduce the features available in the Skyline Targeted Proteomics Environment for refining instrument methods for Selected Reaction Monitoring (SRM,
Package CorrectOverloadedPeaks
 Type Package Package CorrectOverloadedPeaks July 10, 2018 Title Correct Overloaded Peaks from GC-APCI-MS Data Version 1.2.15 Date 2018-07-10 Author Jan Lisec [aut, cre] Analyzes and modifies metabolomics
Type Package Package CorrectOverloadedPeaks July 10, 2018 Title Correct Overloaded Peaks from GC-APCI-MS Data Version 1.2.15 Date 2018-07-10 Author Jan Lisec [aut, cre] Analyzes and modifies metabolomics
Progenesis QI for proteomics User Guide. Analysis workflow guidelines for DDA data
 Progenesis QI for proteomics User Guide Analysis workflow guidelines for DDA data Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using Progenesis QI for proteomics?...
Progenesis QI for proteomics User Guide Analysis workflow guidelines for DDA data Contents Introduction... 3 How to use this document... 3 How can I analyse my own runs using Progenesis QI for proteomics?...
Tutorials. Saturn GC/MS Workstation Version 5.4. Varian Analytical Instruments 2700 Mitchell Drive Walnut Creek, CA /usa
 Varian Analytical Instruments 2700 Mitchell Drive Walnut Creek, CA 94598-1675/usa Saturn GC/MS Workstation Version 5.4 Tutorials Varian, Inc. 1999 03-914765-00:Rev. 1 1 Contents How to Exercises...1 Overview...1
Varian Analytical Instruments 2700 Mitchell Drive Walnut Creek, CA 94598-1675/usa Saturn GC/MS Workstation Version 5.4 Tutorials Varian, Inc. 1999 03-914765-00:Rev. 1 1 Contents How to Exercises...1 Overview...1
NMR Users Guide Organic Chemistry Laboratory
 NMR Users Guide Organic Chemistry Laboratory Introduction The chemistry department is fortunate to have a high field (400 MHz) Nuclear Magnetic Resonance (NMR) spectrometer. You will be using this instrument
NMR Users Guide Organic Chemistry Laboratory Introduction The chemistry department is fortunate to have a high field (400 MHz) Nuclear Magnetic Resonance (NMR) spectrometer. You will be using this instrument
Mnova Training Basics
 Mnova Training Basics Version 12.0.3 Oct. 23, 2018 Chen Peng, PhD, VP of Business Development, North America & Asia Mestrelab Research SL chen.peng@mestrelab.com 858.736.4563 Main Topics Installation and
Mnova Training Basics Version 12.0.3 Oct. 23, 2018 Chen Peng, PhD, VP of Business Development, North America & Asia Mestrelab Research SL chen.peng@mestrelab.com 858.736.4563 Main Topics Installation and
Xcalibur. QuickQuan. User Guide. XCALI Revision C July For Research Use Only Not for use in Diagnostic Procedures
 Xcalibur QuickQuan User Guide XCALI-97195 Revision C July 2007 For Research Use Only Not for use in Diagnostic Procedures 2007 Thermo Fisher Scientific Inc. All rights reserved. Microsoft, Access, Excel,
Xcalibur QuickQuan User Guide XCALI-97195 Revision C July 2007 For Research Use Only Not for use in Diagnostic Procedures 2007 Thermo Fisher Scientific Inc. All rights reserved. Microsoft, Access, Excel,
TraceFinder Administrator Quick Reference Guide
 TraceFinder Administrator Quick Reference Guide This quick reference guide discusses the application configuration tasks assigned to the ITAdmin and LabDirector roles when user security is activated in
TraceFinder Administrator Quick Reference Guide This quick reference guide discusses the application configuration tasks assigned to the ITAdmin and LabDirector roles when user security is activated in
PEAKS Studio 5.1 User s Manual
 BIOINFORMATICS SOLUTIONS INC. PEAKS Studio 5.1 User s Manual Bioinformatics Solutions Inc. 470 Weber St. N. Suite 204 Waterloo, Ontario, Canada N2L 6J2 Phone 519-885-8288 Fax 519-885-9075 Please contact
BIOINFORMATICS SOLUTIONS INC. PEAKS Studio 5.1 User s Manual Bioinformatics Solutions Inc. 470 Weber St. N. Suite 204 Waterloo, Ontario, Canada N2L 6J2 Phone 519-885-8288 Fax 519-885-9075 Please contact
PEAKS Studio 5 User s Manual
 BIOINFORMATICS SOLUTIONS INC PEAKS Studio 5 User s Manual Bioinformatics Solutions Inc. 470 Weber St. N. Suite 204 Waterloo, Ontario, Canada N2L 6J2 Phone 519-885-8288 Fax 519-885-9075 Please contact BSI
BIOINFORMATICS SOLUTIONS INC PEAKS Studio 5 User s Manual Bioinformatics Solutions Inc. 470 Weber St. N. Suite 204 Waterloo, Ontario, Canada N2L 6J2 Phone 519-885-8288 Fax 519-885-9075 Please contact BSI
Package HiResTEC. August 7, 2018
 Type Package Package HiResTEC August 7, 2018 Title Non-Targeted Fluxomics on High-Resolution Mass-Spectrometry Data Version 0.54 Date 2018-08-07 Maintainer Jan Lisec Identifying labeled
Type Package Package HiResTEC August 7, 2018 Title Non-Targeted Fluxomics on High-Resolution Mass-Spectrometry Data Version 0.54 Date 2018-08-07 Maintainer Jan Lisec Identifying labeled
Retention Time Locking with the MSD Productivity ChemStation. Technical Overview. Introduction. When Should I Lock My Methods?
 Retention Time Locking with the MSD Productivity ChemStation Technical Overview Introduction A retention time is the fundamental qualitative measurement of chromatography. Most peak identification is performed
Retention Time Locking with the MSD Productivity ChemStation Technical Overview Introduction A retention time is the fundamental qualitative measurement of chromatography. Most peak identification is performed
REVIEW. MassFinder 3. Navigation of the Chromatogram
 REVIEW MassFinder 3 Dr. Detlev H. Hochmuth, Author Hochmuth Scientific Consulting Hamburg, Germany http://www.massfinder.com Euro 1850, which includes latest version of MassFinder 3, a terpene spectral
REVIEW MassFinder 3 Dr. Detlev H. Hochmuth, Author Hochmuth Scientific Consulting Hamburg, Germany http://www.massfinder.com Euro 1850, which includes latest version of MassFinder 3, a terpene spectral
You will remember from the introduction, that sequence queries are searches where mass information is combined with amino acid sequence or
 1 You will remember from the introduction, that sequence queries are searches where mass information is combined with amino acid sequence or composition information 2 The best known example is a sequence
1 You will remember from the introduction, that sequence queries are searches where mass information is combined with amino acid sequence or composition information 2 The best known example is a sequence
Spectrometer Visible Light Spectrometer V4.4
 Visible Light Spectrometer V4.4 Table of Contents Package Contents...3 Trademarks...4 Manual Driver and Application installation...5 Manual Application Installation...6 First Start of the Application...8
Visible Light Spectrometer V4.4 Table of Contents Package Contents...3 Trademarks...4 Manual Driver and Application installation...5 Manual Application Installation...6 First Start of the Application...8
GPS Explorer Software For Protein Identification Using the Applied Biosystems 4700 Proteomics Analyzer
 GPS Explorer Software For Protein Identification Using the Applied Biosystems 4700 Proteomics Analyzer Getting Started Guide GPS Explorer Software For Protein Identification Using the Applied Biosystems
GPS Explorer Software For Protein Identification Using the Applied Biosystems 4700 Proteomics Analyzer Getting Started Guide GPS Explorer Software For Protein Identification Using the Applied Biosystems
Direct Infusion Mass Spectrometry Processing (DIMaSP) Instructions for use
 Instructions for use") Direct Infusion Mass Spectrometry Processing (DIMaSP) Instructions for use The following instructions assume the user has a batch of paired sample and blank spectra that have been exported to a comma-separated
Direct Infusion Mass Spectrometry Processing (DIMaSP) Instructions for use The following instructions assume the user has a batch of paired sample and blank spectra that have been exported to a comma-separated
Agilent G6855AA MassHunter Personal
 Agilent G6855AA MassHunter Personal Forensic Toxicology Database Kit Quick Start Guide What is the MassHunter Personal Forensic Toxicology Database Kit? 1 Kit Content 2 Where to find more information 4
Agilent G6855AA MassHunter Personal Forensic Toxicology Database Kit Quick Start Guide What is the MassHunter Personal Forensic Toxicology Database Kit? 1 Kit Content 2 Where to find more information 4
To get started download the dataset, unzip files, start Maven, and follow steps below.
 Getting Started. This document provides basic overview of Maven functionality. For the purpose of demonstration we will use an example CMV Viral Infection time course dataset. (Dataset Download). There
Getting Started. This document provides basic overview of Maven functionality. For the purpose of demonstration we will use an example CMV Viral Infection time course dataset. (Dataset Download). There
MassHunter File Reader
 MassHunter File Reader vers 1.0.0 2015 Quadtech Associates, Inc. All Rights Reserved Release date: November 18, 2015 www.quadtechassociates.com MassHunter File Reader Welcome to MassHunter File Reader.
MassHunter File Reader vers 1.0.0 2015 Quadtech Associates, Inc. All Rights Reserved Release date: November 18, 2015 www.quadtechassociates.com MassHunter File Reader Welcome to MassHunter File Reader.
TopSpin. Multiplet Analysis Tutorial
 TopSpin Multiplet Analysis Tutorial Contents Automatic Multiplet Definition Manual Single-Level Multiplet Definition Manual Multi-Level Multiplet Definition Working with Multiplets Report dialog and export
TopSpin Multiplet Analysis Tutorial Contents Automatic Multiplet Definition Manual Single-Level Multiplet Definition Manual Multi-Level Multiplet Definition Working with Multiplets Report dialog and export
TraceFinder Analysis Quick Reference Guide
 TraceFinder Analysis Quick Reference Guide This quick reference guide describes the Analysis mode tasks assigned to the Technician role in Thermo TraceFinder analytical software. For detailed descriptions
TraceFinder Analysis Quick Reference Guide This quick reference guide describes the Analysis mode tasks assigned to the Technician role in Thermo TraceFinder analytical software. For detailed descriptions
This manual describes step-by-step instructions to perform basic operations for data analysis.
 HDXanalyzer User Manual The program HDXanalyzer is available for the analysis of the deuterium exchange mass spectrometry data obtained on high-resolution mass spectrometers. Currently, the program is
HDXanalyzer User Manual The program HDXanalyzer is available for the analysis of the deuterium exchange mass spectrometry data obtained on high-resolution mass spectrometers. Currently, the program is
Agilent G6854AA MassHunter Personal Pesticide Database Kit Quick Start Guide
 Agilent G6854AA MassHunter Personal Pesticide Database Kit Quick Start Guide What is the MassHunter Personal Pesticide Database Kit? 1 Kit Content 2 Where to find more information 3 Before You Begin 4
Agilent G6854AA MassHunter Personal Pesticide Database Kit Quick Start Guide What is the MassHunter Personal Pesticide Database Kit? 1 Kit Content 2 Where to find more information 3 Before You Begin 4
Fatty Acid Methyl Ester (FAME) RTL Databases for GC and GC/MS
 RTL Databases for GC and GC/MS") Fatty Acid Methyl Ester (FAME) RTL Databases for GC and GC/MS User contributed by: Frank David and Pat Sandra Research Institute for Chromatography Pres. Kennedypark 20, B-8500 Kortrijk, Belgium and by:
Fatty Acid Methyl Ester (FAME) RTL Databases for GC and GC/MS User contributed by: Frank David and Pat Sandra Research Institute for Chromatography Pres. Kennedypark 20, B-8500 Kortrijk, Belgium and by:
Chromeleon / MSQ Plus Operator s Guide Document No Revision 03 October 2009
 MSQ Plus Chromeleon / MSQ Plus Operator s Guide Document No. 065322 Revision 03 October 2009 Chromeleon / MSQ Plus Operator s Guide 2009 by Dionex Corporation All rights reserved worldwide. Printed in
MSQ Plus Chromeleon / MSQ Plus Operator s Guide Document No. 065322 Revision 03 October 2009 Chromeleon / MSQ Plus Operator s Guide 2009 by Dionex Corporation All rights reserved worldwide. Printed in
MetCirc: Navigating mass spectral similarity in high-resolution MS/MS metabolomics data
 MetCirc: Navigating mass spectral similarity in high-resolution MS/MS metabolomics data Thomas Naake and Emmanuel Gaquerel Plant Defense Metabolism Centre for Organismal Studies August 30, 2018 1 Introduction
MetCirc: Navigating mass spectral similarity in high-resolution MS/MS metabolomics data Thomas Naake and Emmanuel Gaquerel Plant Defense Metabolism Centre for Organismal Studies August 30, 2018 1 Introduction
Reference Manual MarkerView Software Reference Manual Revision: February, 2010
 Reference Manual MarkerView 1.2.1 Software Reference Manual - 1 - Revision: February, 2010 This document is provided to customers who have purchased AB SCIEX equipment to use in the operation of such AB
Reference Manual MarkerView 1.2.1 Software Reference Manual - 1 - Revision: February, 2010 This document is provided to customers who have purchased AB SCIEX equipment to use in the operation of such AB
Using OPUS to Process Evolved Gas Data (8/12/15 edits highlighted)
") Using OPUS to Process Evolved Gas Data (8/12/15 edits highlighted) Once FTIR data has been acquired for the gases evolved during your DSC/TGA run, you will process using the OPUS software package. Select
Using OPUS to Process Evolved Gas Data (8/12/15 edits highlighted) Once FTIR data has been acquired for the gases evolved during your DSC/TGA run, you will process using the OPUS software package. Select
We are painfully aware that we don't have a good, introductory tutorial for Mascot on our web site. Its something that has come up in discussions
 We are painfully aware that we don't have a good, introductory tutorial for Mascot on our web site. Its something that has come up in discussions many times, and we always resolve to do something but then
We are painfully aware that we don't have a good, introductory tutorial for Mascot on our web site. Its something that has come up in discussions many times, and we always resolve to do something but then
Analysis of GC-MS metabolomics data with metams
 Analysis of GC-MS metabolomics data with metams Ron Wehrens October 30, 2017 1 Introduction Many packages are available for the analysis of data from GC-MS and LC-MS experiments typically, hardware vendors
Analysis of GC-MS metabolomics data with metams Ron Wehrens October 30, 2017 1 Introduction Many packages are available for the analysis of data from GC-MS and LC-MS experiments typically, hardware vendors
High-throughput Processing and Analysis of LC-MS Spectra
 High-throughput Processing and Analysis of LC-MS Spectra By Jianguo Xia (jianguox@ualberta.ca) Last update : 02/05/2012 This tutorial shows how to process and analyze LC-MS spectra using methods provided
High-throughput Processing and Analysis of LC-MS Spectra By Jianguo Xia (jianguox@ualberta.ca) Last update : 02/05/2012 This tutorial shows how to process and analyze LC-MS spectra using methods provided
Steps for LCMS-8040 Triple Quadrupole
 Steps for LCMS-8040 Triple Quadrupole June 2017 Version 2 A. Data Acquisition 1. Check lights on MS. There should be a total of three lights lit by LED. One of them is blinking. Power Blinking Gas Status
Steps for LCMS-8040 Triple Quadrupole June 2017 Version 2 A. Data Acquisition 1. Check lights on MS. There should be a total of three lights lit by LED. One of them is blinking. Power Blinking Gas Status
Tutorial for the PNNL Biodiversity Library Skyline Plugin
 Tutorial for the PNNL Biodiversity Library Skyline Plugin Developed By: Michael Degan, Grant Fujimoto, Lillian Ryadinskiy, Samuel H Payne Contact Information: Samuel Payne (Samuel.Payne@pnnl.gov) Michael
Tutorial for the PNNL Biodiversity Library Skyline Plugin Developed By: Michael Degan, Grant Fujimoto, Lillian Ryadinskiy, Samuel H Payne Contact Information: Samuel Payne (Samuel.Payne@pnnl.gov) Michael
Note: Note: Input: Output: Hit:
 MS/MS search 8.9 i The ms/ms search of GPMAW is based on the public domain search engine X! Tandem. The X! Tandem program is a professional class search engine; Although it is able to perform proteome
MS/MS search 8.9 i The ms/ms search of GPMAW is based on the public domain search engine X! Tandem. The X! Tandem program is a professional class search engine; Although it is able to perform proteome
QuiC 1.0 (Owens) User Manual
 User Manual") QuiC 1.0 (Owens) User Manual 1 Contents 2 General Information... 2 2.1 Computer System Requirements... 2 2.2 Scope of QuiCSoftware... 3 2.3 QuiC... 3 2.4 QuiC Release Features... 3 2.4.1 QuiC 1.0... 3
QuiC 1.0 (Owens) User Manual 1 Contents 2 General Information... 2 2.1 Computer System Requirements... 2 2.2 Scope of QuiCSoftware... 3 2.3 QuiC... 3 2.4 QuiC Release Features... 3 2.4.1 QuiC 1.0... 3
Spectronaut Pulsar X
 Spectronaut Pulsar X User Manual 1 General Information... 8 1.1 Scope of Spectronaut Pulsar X Software... 8 1.2 Spectronaut Pulsar X Release Features... 8 1.3 Computer System Requirements... 9 1.4 Post
Spectronaut Pulsar X User Manual 1 General Information... 8 1.1 Scope of Spectronaut Pulsar X Software... 8 1.2 Spectronaut Pulsar X Release Features... 8 1.3 Computer System Requirements... 9 1.4 Post
A Batch Import Module for an Empirically Derived Mass Spectral Database
 A Batch Import Module for an Empirically Derived Mass Spectral Database Jennifer Van Puymbrouck, 1 David Angulo, 2 Kevin Drew, 3 Lee Ann Hollenbeck, 4 Dominic Battre, 5 Alex Schilling, 6 David Jabon, 7
A Batch Import Module for an Empirically Derived Mass Spectral Database Jennifer Van Puymbrouck, 1 David Angulo, 2 Kevin Drew, 3 Lee Ann Hollenbeck, 4 Dominic Battre, 5 Alex Schilling, 6 David Jabon, 7
Project Report on. De novo Peptide Sequencing. Course: Math 574 Gaurav Kulkarni Washington State University
 Project Report on De novo Peptide Sequencing Course: Math 574 Gaurav Kulkarni Washington State University Introduction Protein is the fundamental building block of one s body. Many biological processes
Project Report on De novo Peptide Sequencing Course: Math 574 Gaurav Kulkarni Washington State University Introduction Protein is the fundamental building block of one s body. Many biological processes
SpectroDive 8 - Coelacanth. User Manual
 SpectroDive 8 - Coelacanth User Manual Table of Contents 1 System Requirements... 4 2 General Information... 4 2.1 Supported Instruments... 4 3 Getting Started... 5 3.1 Getting SpectroDive... 5 3.2 SpectroDive
SpectroDive 8 - Coelacanth User Manual Table of Contents 1 System Requirements... 4 2 General Information... 4 2.1 Supported Instruments... 4 3 Getting Started... 5 3.1 Getting SpectroDive... 5 3.2 SpectroDive
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench
 Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Quick Start Guide What is the Spectrum Mill MS Proteomics Workbench? 2 What s New in Version B.06.00? 3 Where to Find More Information 9 Setting
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Quick Start Guide What is the Spectrum Mill MS Proteomics Workbench? 2 What s New in Version B.06.00? 3 Where to Find More Information 9 Setting
Agilent 6400 Series Triple Quadrupole LC/MS System
 Agilent 6400 Series Triple Quadrupole LC/MS System Quick Start Guide Where to find information 4 Getting Started 6 Step 1. Start the Data Acquisition software 7 Step 2. Prepare the LC modules 13 Step 3.
Agilent 6400 Series Triple Quadrupole LC/MS System Quick Start Guide Where to find information 4 Getting Started 6 Step 1. Start the Data Acquisition software 7 Step 2. Prepare the LC modules 13 Step 3.
An introduction to Baitmet package
 An introduction to Baitmet package Version 1.0.1 Xavier Domingo-Almenara (Maintainer) xdomingo@scripps.edu May 17, 2017 This vignette presents Baitmet, an R package for library driven compound profiling
An introduction to Baitmet package Version 1.0.1 Xavier Domingo-Almenara (Maintainer) xdomingo@scripps.edu May 17, 2017 This vignette presents Baitmet, an R package for library driven compound profiling
NIST/EPA/NIH Mass Spectral Library (NIST 05) and NIST Mass Spectral Search Program (Version 2.0d)
 and NIST Mass Spectral Search Program (Version 2.0d)") NIST Standard Reference Database 1A NIST/EPA/NIH Mass Spectral Library (NIST 05) and NIST Mass Spectral Search Program (Version 2.0d) For Use with Microsoft Windows User s Guide Scientific Instrument Services,
NIST Standard Reference Database 1A NIST/EPA/NIH Mass Spectral Library (NIST 05) and NIST Mass Spectral Search Program (Version 2.0d) For Use with Microsoft Windows User s Guide Scientific Instrument Services,
PROTEOMIC COMMAND LINE SOLUTION. Linux User Guide December, B i. Bioinformatics Solutions Inc.
 >_ PROTEOMIC COMMAND LINE SOLUTION Linux User Guide December, 2015 B i Bioinformatics Solutions Inc. www.bioinfor.com 1. Introduction Liquid chromatography-tandem mass spectrometry (LC-MS/MS) based proteomics
>_ PROTEOMIC COMMAND LINE SOLUTION Linux User Guide December, 2015 B i Bioinformatics Solutions Inc. www.bioinfor.com 1. Introduction Liquid chromatography-tandem mass spectrometry (LC-MS/MS) based proteomics
MultiGlycan ESI User Manual
 MultiGlycan ESI User Manual 1. Download User can download the latest MultiGlycan ESI from our website http://darwin.informatics.indiana.edu/multiglycan/ 2. Installation 2.1. Unzip Unzip downloaded file
MultiGlycan ESI User Manual 1. Download User can download the latest MultiGlycan ESI from our website http://darwin.informatics.indiana.edu/multiglycan/ 2. Installation 2.1. Unzip Unzip downloaded file
SIVIC GUI Overview. SIVIC GUI Layout Overview
 SIVIC GUI Overview SIVIC GUI Layout Overview At the top of the SIVIC GUI is a row of buttons called the Toolbar. It is a quick interface for loading datasets, controlling how the mouse manipulates the
SIVIC GUI Overview SIVIC GUI Layout Overview At the top of the SIVIC GUI is a row of buttons called the Toolbar. It is a quick interface for loading datasets, controlling how the mouse manipulates the
FrAnK: A Web Application for the Annotation of Mass Spectral Peaks Using Fragmentation Spectra
 FrAnK: A Web Application for the Annotation of Mass Spectral Peaks Using Fragmentation Spectra Scott J. Greig School of Computing Science Sir Alwyn Williams Building University of Glasgow G12 8RZ A dissertation
FrAnK: A Web Application for the Annotation of Mass Spectral Peaks Using Fragmentation Spectra Scott J. Greig School of Computing Science Sir Alwyn Williams Building University of Glasgow G12 8RZ A dissertation
MIMA V 1.0. MS IMS Mapper. Peak Identification in Ion mobility Spectrometry
 MIMA V 1.0 MS IMS Mapper Peak Identification in Ion mobility Spectrometry I. What is MIMA? MIMA is a software package for mapping mass-spectrometric (MS) data to corresponding ion-mobility spectrometer
MIMA V 1.0 MS IMS Mapper Peak Identification in Ion mobility Spectrometry I. What is MIMA? MIMA is a software package for mapping mass-spectrometric (MS) data to corresponding ion-mobility spectrometer
Agilent 6300 Ion Trap LC/MS Systems Quick Start Guide
 Agilent 6300 Ion Trap LC/MS Systems Quick Start Guide A guide to help you get started with the Agilent 6300 Series Ion Trap LC/MS system What is the Agilent 6300 Series Ion Trap LC/MS System? 2 Where to
Agilent 6300 Ion Trap LC/MS Systems Quick Start Guide A guide to help you get started with the Agilent 6300 Series Ion Trap LC/MS system What is the Agilent 6300 Series Ion Trap LC/MS System? 2 Where to
User guide. Using the ALEX 123 framework to identify lipid molecules detected by MALDI-based high-resolution FTMS 1 and ITMS 2 analysis
 User guide for Using the ALEX 123 framework to identify lipid molecules detected by MALDI-based high-resolution FTMS 1 and ITMS 2 analysis Used in the publication: Ellis et al. (2018, in review) Version
User guide for Using the ALEX 123 framework to identify lipid molecules detected by MALDI-based high-resolution FTMS 1 and ITMS 2 analysis Used in the publication: Ellis et al. (2018, in review) Version