Multiple Sequence Alignment Gene Finding, Conserved Elements
|
|
|
- Denis Mosley
- 5 years ago
- Views:
Transcription









1 Multiple Sequence Alignment Gene Finding, Conserved Elements

2 Definition Given N sequences x 1, x 2,, x N : Insert gaps (-) in each sequence x i, such that All sequences have the same length L Score of the global map is maximum



3 Applications

4 Gene structure intron1 intron2 exon1 exon2 exon3 transcription splicing translation exon = protein-coding intron = non-coding Codon: A triplet of nucleotides that is converted to one amino acid

5 Gene Finding EXON EXON EXON EXON EXON Classes of Gene predictors Ab initio: Only look at the genomic DNA of target genome De novo: Target genome + aligned informant genome(s) Human tttcttagactttaaagctgtcaagccgtgttctagataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaatttgatgaagtatgtaccta Macaque tttcttagactttaaagctgtcaagccgtgttctagataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaatttgatgaagtatgtaccta Mouse ttgcttagactttaaagttgtcaagccgcgttcttgataaaataagtattggacaacttgttagtcttctttccaacaacctgaacaaatttgatgaagtatgta-cca Rat ttgcttagactttaaagttgtcaagccgtgttcttgataaaataagtattggacaacttattagtcttctttccaacaacctgaacaaatttgatgaagtatgtaccca Rabbit t--attagactttaaagctgtcaagccgtgttctagataaaataagtattgggcaacttattagtctcctttccaacaacctgaacaaatttgatgaagtatgtaccta Dog t-cattagactttaaagctgtcaagccgtgttctggataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaattcgatgaagtatgtaccta Cow t-cattagactttgaagctatcaagccgtgttctggataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaatttgatgaagtatgta-cta Armadillo gca--tagaccttaaaactgtcaagccgtgttttagataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaatttgatgaagtatgtgccta Elephant gct-ttagactttaaaactgtccagccgtgttcttgataaaataagtattggacaacttgtcagtctcctttccaacaacctgaacaaatttgatgaagtatgtatcta Tenrec tc-cttagactttaaaactttcgagccgggttctagataaaataagtattggacaacttgttagtctcctttccaacaacctgaacaaatttgatgaagtatgtatcta Opossum ---tttagaccttaaaactgtcaagccgtgttctagataaaataagcactggacagcttatcagtctcctttccaacaatctgaacaagtttgatgaagtatgtagctg Chicken ----ttagaccttaaaactgtcaagcaaagttctagataaaataagtactggacaattggtcagccttctttccaacaatctgaacaaattcgatgaggtatgtt--tg RNA-seq based approaches Nature Biotechnology 28, (2010)

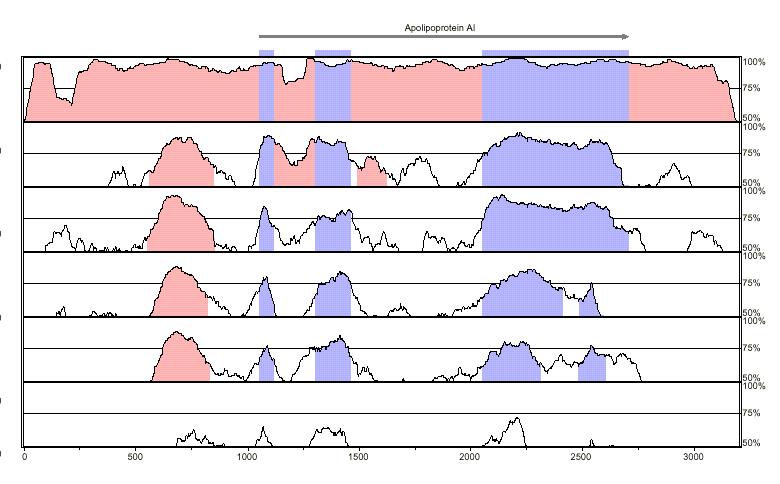



6 Using Comparative Information

7 Patterns of Conservation Genes" Intergenic" Separation" Mutations" Gaps" Frameshifts" 30%" 1.3%" 0.14% " 58%" 14%" 10.2%" à " à " à " 2-fold" 10-fold" 75-fold"
8 Scoring Function: Sum Of Pairs Definition: Induced pairwise alignment A pairwise alignment induced by the multiple alignment Example: Induces: x: AC-GCGG-C y: AC-GC-GAG z: GCCGC-GAG x: ACGCGG-C; x: AC-GCGG-C; y: AC-GCGAG y: ACGC-GAC; z: GCCGC-GAG; z: GCCGCGAG

 = Σ k<l")
9 Sum Of Pairs (cont d) Heuristic way to incorporate evolution tree:.. Weighted SOP: S(m) = Σ k<l w kl s(m k, m l )
10 A Profile Representation - A G G C T A T C A C C T G T A G C T A C C A G C A G C T A C C A G C A G C T A T C A C G G C A G C T A T C G C G G A C G T Given a multiple alignment M = m 1 m n Replace each column m i with profile entry p i Frequency of each letter in Σ # gaps Optional: # gap openings, extensions, closings Can think of this as a likelihood of each letter in each position
11 Multiple Sequence Alignments Algorithms
12 Multidimensional DP Generalization of Needleman-Wunsh: S(m) = Σ i S(m i ) (sum of column scores) F(i 1,i 2,,i N ): Optimal alignment up to (i 1,, i N ) F(i 1,i 2,,i N ) = max (all neighbors of cube) (F(nbr)+S(nbr))
13 Multidimensional DP Example: in 3D (three sequences): 7 neighbors/cell F(i,j,k) = max{ F(i 1, j 1, k 1) + S(x i, x j, x k ), F(i 1, j 1, k ) + S(x i, x j, - ), F(i 1, j, k 1) + S(x i, -, x k ), F(i 1, j, k ) + S(x i, -, - ), F(i, j 1, k 1) + S( -, x j, x k ), F(i, j 1, k ) + S( -, x j, - ), F(i, j, k 1) + S( -, -, x k ) }

14 Multidimensional DP Running Time: 1. Size of matrix: L N ; Where L = length of each sequence N = number of sequences 2. Neighbors/cell: 2 N 1 Therefore O(2 N L N )
15 Progressive Alignment p xyzw p zw p xy x y z w When evolutionary tree is known: Align closest first, in the order of the tree In each step, align two sequences x, y, or profiles p x, p y, to generate a new alignment with associated profile p result Weighted version: Tree edges have weights, proportional to the divergence in that edge New profile is a weighted average of two old profiles
16 Progressive Alignment When evolutionary tree is known: s(p x, p y ) = 0.8*0.6*s(A, A) + 0.2*0.6*s(C, A) + 0.8*0.4*s(A, -) + 0.2*0.4*s(C, -) Align closest first, in the order of the tree In each step, align two sequences Result: x, y, or profiles p p x, p y, to generate a new alignment with associated profile p xy = (0.7, 0.1, 0, 0, 0.2) result Weighted version: s(p x, -) = 0.8*1.0*s(A, -) + 0.2*1.0*s(C, -) Tree edges have weights, proportional to the divergence in that edge Result: p New profile is a weighted average of two old x- = (0.4, 0.1, 0, 0, 0.5) profiles x y Example z Profile: (A, C, G, T, -) p x = (0.8, 0.2, 0, 0, 0) w p y = (0.6, 0, 0, 0, 0.4)
17 Progressive Alignment? x y z w When evolutionary tree is unknown: Perform all pairwise alignments Define distance matrix D, where D(x, y) is a measure of evolutionary distance, based on pairwise alignment Construct a tree (UPGMA / Neighbor Joining / Other methods) Align on the tree
18 Heuristics to improve alignments Iterative refinement schemes A*-based search Consistency Simulated Annealing
19 Iterative Refinement One problem of progressive alignment: Initial alignments are frozen even when new evidence comes Example: x: GAAGTT y: GAC-TT z: GAACTG w: GTACTG Frozen! Now clear correct y = GA-CTT
20 Iterative Refinement Algorithm (Barton-Stenberg): 1. For j = 1 to N, Remove x j, and realign to x 1 x j-1 x j+1 x N 2. Repeat 4 until convergence y z x allow y to vary x,z fixed projection
21 Iterative Refinement Example: align (x,y), (z,w), (xy, zw): x: GAAGTTA y: GAC-TTA z: GAACTGA w: GTACTGA After realigning y: x: GAAGTTA y: G-ACTTA + 3 matches z: GAACTGA w: GTACTGA
22 Iterative Refinement Example not handled well: x: GAAGTTA y 1 : GAC-TTA y 2 : GAC-TTA y 3 : GAC-TTA Realigning any single y i changes nothing z: GAACTGA w: GTACTGA
23 Consistency z z k x x i y y j y j
24 Consistency z z k x x i y y j y j Basic method for applying consistency Compute all pairs of alignments xy, xz, yz, When aligning x, y during progressive alignment, For each (x i, y j ), let s(x i, y j ) = function_of(x i, y j, a xz, a yz ) Align x and y with DP using the modified s(.,.) function
25 Real-world protein aligners MUSCLE High throughput One of the best in accuracy ProbCons High accuracy Reasonable speed
26 MUSCLE at a glance 1. Fast measurement of all pairwise distances between sequences D DRAFT (x, y) defined in terms of # common k-mers (k~3) O(N 2 L logl) time 2. Build tree T DRAFT based on those distances, with UPGMA 3. Progressive alignment over T DRAFT, resulting in multiple alignment M DRAFT 4. Measure new Kimura-based distances D(x, y) based on M DRAFT 5. Build tree T based on D 6. Progressive alignment over T, to build M 7. Iterative refinement; for many rounds, do: Tree Partitioning: Split M on one branch and realign the two resulting profiles If new alignment M has better sum-of-pairs score than previous one, accept
27 PROBCONS at a glance 1. Computation of all posterior matrices M xy : M xy (i, j) = Prob(x i ~ y j ), using a HMM 2. Re-estimation of posterior matrices M xy with probabilistic consistency M xy (i, j) = 1/N Σ sequence z Σ k M xz (i, k) M yz (j, k); M xy = Avg z (M xz M zy ) 3. Compute for every pair x, y, the maximum expected accuracy alignment A xy : alignment that maximizes Σ aligned (i, j) in A M xy (i, j) Define E(x, y) = Σ aligned (i, j) in Axy M xy (i, j) 4. Build tree T with hierarchical clustering using similarity measure E(x, y) 5. Progressive alignment on T to maximize E(.,.) 6. Iterative refinement; for many rounds, do: Randomized Partitioning: Split sequences in M in two subsets by flipping a coin for each sequence and realign the two resulting profiles


28 Mammalian alignments References Lindblad-Toh et al. Nature 478: , 2011 Lin et al. Genome Research 21: , 2011
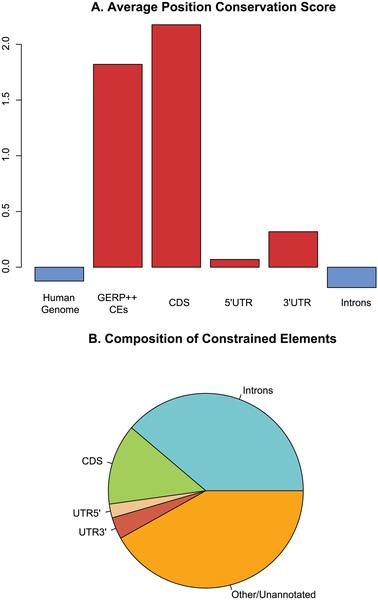

29 Genome Evolutionary Rate Profiling (GERP)
Multiple Sequence Alignment. With thanks to Eric Stone and Steffen Heber, North Carolina State University
 Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004
 CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004 Lecture #4: 8 April 2004 Topics: Sequence Similarity Scribe: Sonil Mukherjee 1 Introduction
CS273: Algorithms for Structure Handout # 4 and Motion in Biology Stanford University Thursday, 8 April 2004 Lecture #4: 8 April 2004 Topics: Sequence Similarity Scribe: Sonil Mukherjee 1 Introduction
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment
 Multiple Sequence Alignment") GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
GLOBEX Bioinformatics (Summer 2015) Multiple Sequence Alignment Scoring Dynamic Programming algorithms Heuristic algorithms CLUSTAL W Courtesy of jalview Motivations Collective (or aggregate) statistic
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
Alignment ABC. Most slides are modified from Serafim s lectures
 Alignment ABC Most slides are modified from Serafim s lectures Complete genomes Evolution Evolution at the DNA level C ACGGTGCAGTCACCA ACGTTGCAGTCCACCA SEQUENCE EDITS REARRANGEMENTS Sequence conservation
Alignment ABC Most slides are modified from Serafim s lectures Complete genomes Evolution Evolution at the DNA level C ACGGTGCAGTCACCA ACGTTGCAGTCCACCA SEQUENCE EDITS REARRANGEMENTS Sequence conservation
PROTEIN MULTIPLE ALIGNMENT MOTIVATION: BACKGROUND: Marina Sirota
 Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
MULTIPLE SEQUENCE ALIGNMENT
 MULTIPLE SEQUENCE ALIGNMENT Multiple Alignment versus Pairwise Alignment Up until now we have only tried to align two sequences. What about more than two? A faint similarity between two sequences becomes
MULTIPLE SEQUENCE ALIGNMENT Multiple Alignment versus Pairwise Alignment Up until now we have only tried to align two sequences. What about more than two? A faint similarity between two sequences becomes
LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA
 LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA Michael Brudno, Chuong B. Do, Gregory M. Cooper, et al. Presented by Xuebei Yang About Alignments Pairwise Alignments
LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA Michael Brudno, Chuong B. Do, Gregory M. Cooper, et al. Presented by Xuebei Yang About Alignments Pairwise Alignments
Multiple Sequence Alignment II
 Multiple Sequence Alignment II Lectures 20 Dec 5, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline
Multiple Sequence Alignment II Lectures 20 Dec 5, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline
Lecture 10: Local Alignments
 Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Lecture 10: Local Alignments Study Chapter 6.8-6.10 1 Outline Edit Distances Longest Common Subsequence Global Sequence Alignment Scoring Matrices Local Sequence Alignment Alignment with Affine Gap Penalties
Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment
 Scoring Multiple Alignments Entropy Sum of Pairs Alignment") Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment Partial Order Alignment (POA) A-Bruijin (ABA) Approach
Dynamic Programming in 3-D Progressive Alignment Profile Progressive Alignment (ClustalW) Scoring Multiple Alignments Entropy Sum of Pairs Alignment Partial Order Alignment (POA) A-Bruijin (ABA) Approach
Stephen Scott.
 1 / 33 sscott@cse.unl.edu 2 / 33 Start with a set of sequences In each column, residues are homolgous Residues occupy similar positions in 3D structure Residues diverge from a common ancestral residue
1 / 33 sscott@cse.unl.edu 2 / 33 Start with a set of sequences In each column, residues are homolgous Residues occupy similar positions in 3D structure Residues diverge from a common ancestral residue
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment
 Multiple Sequence Alignment") CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Genome 559: Introduction to Statistical and Computational Genomics. Lecture15a Multiple Sequence Alignment Larry Ruzzo
 Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Genome 559: Introduction to Statistical and Computational Genomics Lecture15a Multiple Sequence Alignment Larry Ruzzo 1 Multiple Alignment: Motivations Common structure, function, or origin may be only
Lecture 5: Markov models
 Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Quiz Section Week 8 May 17, Machine learning and Support Vector Machines
 Quiz Section Week 8 May 17, 2016 Machine learning and Support Vector Machines Another definition of supervised machine learning Given N training examples (objects) {(x 1,y 1 ), (x 2,y 2 ),, (x N,y N )}
Quiz Section Week 8 May 17, 2016 Machine learning and Support Vector Machines Another definition of supervised machine learning Given N training examples (objects) {(x 1,y 1 ), (x 2,y 2 ),, (x N,y N )}
Multiple Sequence Alignment (MSA)
") I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
I519 Introduction to Bioinformatics, Fall 2013 Multiple Sequence Alignment (MSA) Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Outline Multiple sequence alignment (MSA) Generalize
Recent Research Results. Evolutionary Trees Distance Methods
 Recent Research Results Evolutionary Trees Distance Methods Indo-European Languages After Tandy Warnow What is the purpose? Understand evolutionary history (relationship between species). Uderstand how
Recent Research Results Evolutionary Trees Distance Methods Indo-European Languages After Tandy Warnow What is the purpose? Understand evolutionary history (relationship between species). Uderstand how
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
A multiple alignment tool in 3D
 Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
Codon models. In reality we use codon model Amino acid substitution rates meet nucleotide models Codon(nucleotide triplet)
") Phylogeny Codon models Last lecture: poor man s way of calculating dn/ds (Ka/Ks) Tabulate synonymous/non- synonymous substitutions Normalize by the possibilities Transform to genetic distance K JC or K
Phylogeny Codon models Last lecture: poor man s way of calculating dn/ds (Ka/Ks) Tabulate synonymous/non- synonymous substitutions Normalize by the possibilities Transform to genetic distance K JC or K
Basics of Multiple Sequence Alignment
 Basics of Multiple Sequence Alignment Tandy Warnow February 10, 2018 Basics of Multiple Sequence Alignment Tandy Warnow Basic issues What is a multiple sequence alignment? Evolutionary processes operating
Basics of Multiple Sequence Alignment Tandy Warnow February 10, 2018 Basics of Multiple Sequence Alignment Tandy Warnow Basic issues What is a multiple sequence alignment? Evolutionary processes operating
Lecture 5: Multiple sequence alignment
 Lecture 5: Multiple sequence alignment Introduction to Computational Biology Teresa Przytycka, PhD (with some additions by Martin Vingron) Why do we need multiple sequence alignment Pairwise sequence alignment
Lecture 5: Multiple sequence alignment Introduction to Computational Biology Teresa Przytycka, PhD (with some additions by Martin Vingron) Why do we need multiple sequence alignment Pairwise sequence alignment
Gribskov Profile. Hidden Markov Models. Building a Hidden Markov Model #$ %&
 Gribskov Profile #$ %& Hidden Markov Models Building a Hidden Markov Model "! Proteins, DNA and other genomic features can be classified into families of related sequences and structures How to detect
Gribskov Profile #$ %& Hidden Markov Models Building a Hidden Markov Model "! Proteins, DNA and other genomic features can be classified into families of related sequences and structures How to detect
Eval: A Gene Set Comparison System
 Masters Project Report Eval: A Gene Set Comparison System Evan Keibler evan@cse.wustl.edu Table of Contents Table of Contents... - 2 - Chapter 1: Introduction... - 5-1.1 Gene Structure... - 5-1.2 Gene
Masters Project Report Eval: A Gene Set Comparison System Evan Keibler evan@cse.wustl.edu Table of Contents Table of Contents... - 2 - Chapter 1: Introduction... - 5-1.1 Gene Structure... - 5-1.2 Gene
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Profiles and Multiple Alignments. COMP 571 Luay Nakhleh, Rice University
 Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Evolutionary tree reconstruction (Chapter 10)
") Evolutionary tree reconstruction (Chapter 10) Early Evolutionary Studies Anatomical features were the dominant criteria used to derive evolutionary relationships between species since Darwin till early
Evolutionary tree reconstruction (Chapter 10) Early Evolutionary Studies Anatomical features were the dominant criteria used to derive evolutionary relationships between species since Darwin till early
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Alignment of Pairs of Sequences
 Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace.
 5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
Sequence clustering. Introduction. Clustering basics. Hierarchical clustering
 Sequence clustering Introduction Data clustering is one of the key tools used in various incarnations of data-mining - trying to make sense of large datasets. It is, thus, natural to ask whether clustering
Sequence clustering Introduction Data clustering is one of the key tools used in various incarnations of data-mining - trying to make sense of large datasets. It is, thus, natural to ask whether clustering
Biology 644: Bioinformatics
 A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
A statistical Markov model in which the system being modeled is assumed to be a Markov process with unobserved (hidden) states in the training data. First used in speech and handwriting recognition In
Chapter 6. Multiple sequence alignment (week 10)
") Course organization Introduction ( Week 1,2) Part I: Algorithms for Sequence Analysis (Week 1-11) Chapter 1-3, Models and theories» Probability theory and Statistics (Week 3)» Algorithm complexity analysis
Course organization Introduction ( Week 1,2) Part I: Algorithms for Sequence Analysis (Week 1-11) Chapter 1-3, Models and theories» Probability theory and Statistics (Week 3)» Algorithm complexity analysis
Chapter 8 Multiple sequence alignment. Chaochun Wei Spring 2018
 1896 1920 1987 2006 Chapter 8 Multiple sequence alignment Chaochun Wei Spring 2018 Contents 1. Reading materials 2. Multiple sequence alignment basic algorithms and tools how to improve multiple alignment
1896 1920 1987 2006 Chapter 8 Multiple sequence alignment Chaochun Wei Spring 2018 Contents 1. Reading materials 2. Multiple sequence alignment basic algorithms and tools how to improve multiple alignment
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Multiple sequence alignment. November 20, 2018
 Multiple sequence alignment November 20, 2018 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one
Multiple sequence alignment November 20, 2018 Why do multiple alignment? Gain insight into evolutionary history Can assess time of divergence by looking at the number of mutations needed to change one
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences
 Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences Yue Lu and Sing-Hoi Sze RECOMB 2007 Presented by: Wanxing Xu March 6, 2008 Content Biology Motivation Computation Problem
Multiple Sequence Alignment Based on Profile Alignment of Intermediate Sequences Yue Lu and Sing-Hoi Sze RECOMB 2007 Presented by: Wanxing Xu March 6, 2008 Content Biology Motivation Computation Problem
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS
 MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS By XU ZHANG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE
MULTIPLE SEQUENCE ALIGNMENT SOLUTIONS AND APPLICATIONS By XU ZHANG A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE
Multiple Sequence Alignment: Multidimensional. Biological Motivation
 Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
BMI/CS 576 Fall 2015 Midterm Exam
 BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
Multiple Sequence Alignment. Mark Whitsitt - NCSA
 Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
Multiple Sequence Alignment Mark Whitsitt - NCSA What is a Multiple Sequence Alignment (MA)? GMHGTVYANYAVDSSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKQPHV GMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHV
1. R. Durbin, S. Eddy, A. Krogh und G. Mitchison: Biological sequence analysis, Cambridge, 1998
 7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Machine Learning. Computational biology: Sequence alignment and profile HMMs
 10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods
 Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods Khaddouja Boujenfa, Nadia Essoussi, and Mohamed Limam International Science Index, Computer and Information Engineering waset.org/publication/482
Comparison of Phylogenetic Trees of Multiple Protein Sequence Alignment Methods Khaddouja Boujenfa, Nadia Essoussi, and Mohamed Limam International Science Index, Computer and Information Engineering waset.org/publication/482
HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT
 HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT - Swarbhanu Chatterjee. Hidden Markov models are a sophisticated and flexible statistical tool for the study of protein models. Using HMMs to analyze proteins
HIDDEN MARKOV MODELS AND SEQUENCE ALIGNMENT - Swarbhanu Chatterjee. Hidden Markov models are a sophisticated and flexible statistical tool for the study of protein models. Using HMMs to analyze proteins
Phylogenetics. Introduction to Bioinformatics Dortmund, Lectures: Sven Rahmann. Exercises: Udo Feldkamp, Michael Wurst
 Phylogenetics Introduction to Bioinformatics Dortmund, 16.-20.07.2007 Lectures: Sven Rahmann Exercises: Udo Feldkamp, Michael Wurst 1 Phylogenetics phylum = tree phylogenetics: reconstruction of evolutionary
Phylogenetics Introduction to Bioinformatics Dortmund, 16.-20.07.2007 Lectures: Sven Rahmann Exercises: Udo Feldkamp, Michael Wurst 1 Phylogenetics phylum = tree phylogenetics: reconstruction of evolutionary
Accelerating the Prediction of Protein Interactions
 Accelerating the Prediction of Protein Interactions Alex Rodionov, Jonathan Rose, Elisabeth R.M. Tillier, Alexandr Bezginov October 21 21 Motivation The human genome is sequenced, but we don't know what
Accelerating the Prediction of Protein Interactions Alex Rodionov, Jonathan Rose, Elisabeth R.M. Tillier, Alexandr Bezginov October 21 21 Motivation The human genome is sequenced, but we don't know what
!"#$ Gribskov Profile. Hidden Markov Models. Building an Hidden Markov Model. Proteins, DNA and other genomic features can be
 Gribskov Profile $ Hidden Markov Models Building an Hidden Markov Model $ Proteins, DN and other genomic features can be classified into families of related sequences and structures $ Related sequences
Gribskov Profile $ Hidden Markov Models Building an Hidden Markov Model $ Proteins, DN and other genomic features can be classified into families of related sequences and structures $ Related sequences
Multiple Sequence Alignment
 Multiple Sequence Alignment Reference: Gusfield, Algorithms on Strings, Trees & Sequences Some slides from: Jones, Pevzner, USC Intro to Bioinformatics Algorithms http://www.bioalgorithms.info/ S. Batzoglu,
Multiple Sequence Alignment Reference: Gusfield, Algorithms on Strings, Trees & Sequences Some slides from: Jones, Pevzner, USC Intro to Bioinformatics Algorithms http://www.bioalgorithms.info/ S. Batzoglu,
1. R. Durbin, S. Eddy, A. Krogh und G. Mitchison: Biological sequence analysis, Cambridge, 1998
 7 Multiple Sequence Alignment The exposition was prepared by Clemens Gröpl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
7 Multiple Sequence Alignment The exposition was prepared by Clemens Gröpl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
Clustering. RNA-seq: What is it good for? Finding Similarly Expressed Genes. Data... And Lots of It!
 RNA-seq: What is it good for? Clustering High-throughput RNA sequencing experiments (RNA-seq) offer the ability to measure simultaneously the expression level of thousands of genes in a single experiment!
RNA-seq: What is it good for? Clustering High-throughput RNA sequencing experiments (RNA-seq) offer the ability to measure simultaneously the expression level of thousands of genes in a single experiment!
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding. B. Majoros M. Pertea S.L. Salzberg
 Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
HMMConverter A tool-box for hidden Markov models with two novel, memory efficient parameter training algorithms
 HMMConverter A tool-box for hidden Markov models with two novel, memory efficient parameter training algorithms by TIN YIN LAM B.Sc., The Chinese University of Hong Kong, 2006 A THESIS SUBMITTED IN PARTIAL
HMMConverter A tool-box for hidden Markov models with two novel, memory efficient parameter training algorithms by TIN YIN LAM B.Sc., The Chinese University of Hong Kong, 2006 A THESIS SUBMITTED IN PARTIAL
Parsimony-Based Approaches to Inferring Phylogenetic Trees
 Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Multiple Sequence Alignment Sum-of-Pairs and ClustalW. Ulf Leser
 Multiple Sequence Alignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence Alignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Multiple Sequence Alignment Sum-of-Pairs and ClustalW Ulf Leser This Lecture Multiple Sequence Alignment The problem Theoretical approach: Sum-of-Pairs scores Practical approach: ClustalW Ulf Leser: Bioinformatics,
Tutorial: chloroplast genomes
 Tutorial: chloroplast genomes Stacia Wyman Department of Computer Sciences Williams College Williamstown, MA 01267 March 10, 2005 ASSUMPTIONS: You are using Internet Explorer under OS X on the Mac. You
Tutorial: chloroplast genomes Stacia Wyman Department of Computer Sciences Williams College Williamstown, MA 01267 March 10, 2005 ASSUMPTIONS: You are using Internet Explorer under OS X on the Mac. You
ChromHMM: automating chromatin-state discovery and characterization
 Nature Methods ChromHMM: automating chromatin-state discovery and characterization Jason Ernst & Manolis Kellis Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure 3 Supplementary Figure
Nature Methods ChromHMM: automating chromatin-state discovery and characterization Jason Ernst & Manolis Kellis Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure 3 Supplementary Figure
Multiple Sequence Alignment Augmented by Expert User Constraints
 Multiple Sequence Alignment Augmented by Expert User Constraints A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of
Multiple Sequence Alignment Augmented by Expert User Constraints A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of
CLC Phylogeny Module User manual
 CLC Phylogeny Module User manual User manual for Phylogeny Module 1.0 Windows, Mac OS X and Linux September 13, 2013 This software is for research purposes only. CLC bio Silkeborgvej 2 Prismet DK-8000
CLC Phylogeny Module User manual User manual for Phylogeny Module 1.0 Windows, Mac OS X and Linux September 13, 2013 This software is for research purposes only. CLC bio Silkeborgvej 2 Prismet DK-8000
Tutorial 1: Exploring the UCSC Genome Browser
 Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS
 INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS MUHANNAD A. ABU-HASHEM, NUR'AINI ABDUL RASHID, ROSNI ABDULLAH, AWSAN A. HASAN AND ATHEER A. ABDULRAZZAQ School of Computer Sciences, Universiti
INVESTIGATION STUDY: AN INTENSIVE ANALYSIS FOR MSA LEADING METHODS MUHANNAD A. ABU-HASHEM, NUR'AINI ABDUL RASHID, ROSNI ABDULLAH, AWSAN A. HASAN AND ATHEER A. ABDULRAZZAQ School of Computer Sciences, Universiti
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
Long Read RNA-seq Mapper
 UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
UNIVERSITY OF ZAGREB FACULTY OF ELECTRICAL ENGENEERING AND COMPUTING MASTER THESIS no. 1005 Long Read RNA-seq Mapper Josip Marić Zagreb, February 2015. Table of Contents 1. Introduction... 1 2. RNA Sequencing...
What is a phylogenetic tree? Algorithms for Computational Biology. Phylogenetics Summary. Di erent types of phylogenetic trees
 What is a phylogenetic tree? Algorithms for Computational Biology Zsuzsanna Lipták speciation events Masters in Molecular and Medical Biotechnology a.a. 25/6, fall term Phylogenetics Summary wolf cat lion
What is a phylogenetic tree? Algorithms for Computational Biology Zsuzsanna Lipták speciation events Masters in Molecular and Medical Biotechnology a.a. 25/6, fall term Phylogenetics Summary wolf cat lion
11/17/2009 Comp 590/Comp Fall
 Lecture 20: Clustering and Evolution Study Chapter 10.4 10.8 Problem Set #5 will be available tonight 11/17/2009 Comp 590/Comp 790-90 Fall 2009 1 Clique Graphs A clique is a graph with every vertex connected
Lecture 20: Clustering and Evolution Study Chapter 10.4 10.8 Problem Set #5 will be available tonight 11/17/2009 Comp 590/Comp 790-90 Fall 2009 1 Clique Graphs A clique is a graph with every vertex connected
Eukaryotic Gene Finding: The GENSCAN System
 Eukaryotic Gene Finding: The GENSCAN System BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC
Eukaryotic Gene Finding: The GENSCAN System BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2016 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC
BMI/CS Lecture #22 - Stochastic Context Free Grammars for RNA Structure Modeling. Colin Dewey (adapted from slides by Mark Craven)
") BMI/CS Lecture #22 - Stochastic Context Free Grammars for RNA Structure Modeling Colin Dewey (adapted from slides by Mark Craven) 2007.04.12 1 Modeling RNA with Stochastic Context Free Grammars consider
BMI/CS Lecture #22 - Stochastic Context Free Grammars for RNA Structure Modeling Colin Dewey (adapted from slides by Mark Craven) 2007.04.12 1 Modeling RNA with Stochastic Context Free Grammars consider
Using multiple alignments to improve seeded local alignment algorithms
 Published online August 12, 2005 Using multiple alignments to improve seeded local alignment algorithms Jason Flannick* and Serafim Batzoglou Department of Computer Science, Stanford University, Stanford,
Published online August 12, 2005 Using multiple alignments to improve seeded local alignment algorithms Jason Flannick* and Serafim Batzoglou Department of Computer Science, Stanford University, Stanford,
Tutorial: RNA-Seq analysis part I: Getting started
 : RNA-Seq analysis part I: Getting started August 9, 2012 CLC bio Finlandsgade 10-12 8200 Aarhus N Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com support@clcbio.com : RNA-Seq analysis
: RNA-Seq analysis part I: Getting started August 9, 2012 CLC bio Finlandsgade 10-12 8200 Aarhus N Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com support@clcbio.com : RNA-Seq analysis
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Using the UCSC genome browser
 Using the UCSC genome browser Credits Terry Braun Mary Mangan, Ph.D. www.openhelix.com UCSC Genome Browser Credits Development team: http://genome.ucsc.edu/staff.html n Led by David Haussler and Jim Kent
Using the UCSC genome browser Credits Terry Braun Mary Mangan, Ph.D. www.openhelix.com UCSC Genome Browser Credits Development team: http://genome.ucsc.edu/staff.html n Led by David Haussler and Jim Kent
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
Lab 4: Multiple Sequence Alignment (MSA)
") Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Olivier Gascuel Arbres formels et Arbre de la Vie Conférence ENS Cachan, septembre Arbres formels et Arbre de la Vie.
 Arbres formels et Arbre de la Vie Olivier Gascuel Centre National de la Recherche Scientifique LIRMM, Montpellier, France www.lirmm.fr/gascuel 10 permanent researchers 2 technical staff 3 postdocs, 10
Arbres formels et Arbre de la Vie Olivier Gascuel Centre National de la Recherche Scientifique LIRMM, Montpellier, France www.lirmm.fr/gascuel 10 permanent researchers 2 technical staff 3 postdocs, 10
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
3.4 Multiple sequence alignment
 3.4 Multiple sequence alignment Why produce a multiple sequence alignment? Using more than two sequences results in a more convincing alignment by revealing conserved regions in ALL of the sequences Aligned
3.4 Multiple sequence alignment Why produce a multiple sequence alignment? Using more than two sequences results in a more convincing alignment by revealing conserved regions in ALL of the sequences Aligned
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
CLUSTERING IN BIOINFORMATICS
 CLUSTERING IN BIOINFORMATICS CSE/BIMM/BENG 8 MAY 4, 0 OVERVIEW Define the clustering problem Motivation: gene expression and microarrays Types of clustering Clustering algorithms Other applications of
CLUSTERING IN BIOINFORMATICS CSE/BIMM/BENG 8 MAY 4, 0 OVERVIEW Define the clustering problem Motivation: gene expression and microarrays Types of clustering Clustering algorithms Other applications of
MSCBIO 2070/02-710: Computational Genomics, Spring A4: spline, HMM, clustering, time-series data analysis, RNA-folding
 MSCBIO 2070/02-710:, Spring 2015 A4: spline, HMM, clustering, time-series data analysis, RNA-folding Due: April 13, 2015 by email to Silvia Liu (silvia.shuchang.liu@gmail.com) TA in charge: Silvia Liu
MSCBIO 2070/02-710:, Spring 2015 A4: spline, HMM, clustering, time-series data analysis, RNA-folding Due: April 13, 2015 by email to Silvia Liu (silvia.shuchang.liu@gmail.com) TA in charge: Silvia Liu
Fast-track to Gene Annotation and Genome Analysis
 Fast-track to Gene Annotation and Genome Analysis Contents Section Page 1.1 Introduction DNA Subway is a bioinformatics workspace that wraps high-level analysis tools in an intuitive and appealing interface.
Fast-track to Gene Annotation and Genome Analysis Contents Section Page 1.1 Introduction DNA Subway is a bioinformatics workspace that wraps high-level analysis tools in an intuitive and appealing interface.
Molecular Evolution & Phylogenetics Complexity of the search space, distance matrix methods, maximum parsimony
 Molecular Evolution & Phylogenetics Complexity of the search space, distance matrix methods, maximum parsimony Basic Bioinformatics Workshop, ILRI Addis Ababa, 12 December 2017 Learning Objectives understand
Molecular Evolution & Phylogenetics Complexity of the search space, distance matrix methods, maximum parsimony Basic Bioinformatics Workshop, ILRI Addis Ababa, 12 December 2017 Learning Objectives understand
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 8: Multiple alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
New String Kernels for Biosequence Data
 Workshop on Kernel Methods in Bioinformatics New String Kernels for Biosequence Data Christina Leslie Department of Computer Science Columbia University Biological Sequence Classification Problems Protein
Workshop on Kernel Methods in Bioinformatics New String Kernels for Biosequence Data Christina Leslie Department of Computer Science Columbia University Biological Sequence Classification Problems Protein
human chimp mouse rat
 Michael rudno These notes are based on earlier notes by Tomas abak Phylogenetic Trees Phylogenetic Trees demonstrate the amoun of evolution, and order of divergence for several genomes. Phylogenetic trees
Michael rudno These notes are based on earlier notes by Tomas abak Phylogenetic Trees Phylogenetic Trees demonstrate the amoun of evolution, and order of divergence for several genomes. Phylogenetic trees
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic