Sequence Alignment & Search
|
|
|
- Mark Montgomery
- 5 years ago
- Views:
Transcription


1 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version of these slides.
2 Lecture Overview Goals: Understand pairwise sequence alignment algorithms Be able to utilize tools for sequence search based on alignments Motivations: Basis for retrieval of sequence-indexed database information Similarity among genomic (amino acid) sequences is a core indicator of homology
3 Part 1: Background
4 Genomic Databases Gene and gene product (e.g. protein) databases are often organized by sequence Genomic sequence encodes all traits of an organism. Gene products are uniquely described by their sequences. Similar sequences among biomolecules indicates both similar function and an evolutionary relationship A located sequence feature (place on a chromosome) is unambiguous and biologically meaningful Closely related to the molecular concept of a gene. => Biologically meaningful database keys
5 Searching sequence databases There are large sequence databases available NCBI Entrez Gene, UniProt Starting from a sequence alone, find information about it Many kinds & sources of input sequences Genomic, expressed, protein (amino acid vs. nucleic acid) Complete or fragmentary sequences Goal is to retrieve a set of similar sequences. Exact matches are rare, and not always interesting Both small differences (mutations) and large (not required for function) within similar sequences can be biologically important.
6 Sequence search & alignment Database organization is focused on efficiency Sequence search doesn t match the traditional database model perfectly Alternative: Start with dynamic programming (a central idea in computational biology) Then explore approximations to it (BLAST)
7 Homology Homology is an evolutionary relationship that either exists or does not. It cannot be partial. An ortholog is a homolog with shared function. A paralog is a homolog that arose through a gene duplication event. Paralogs often have divergent function.
8 Homology
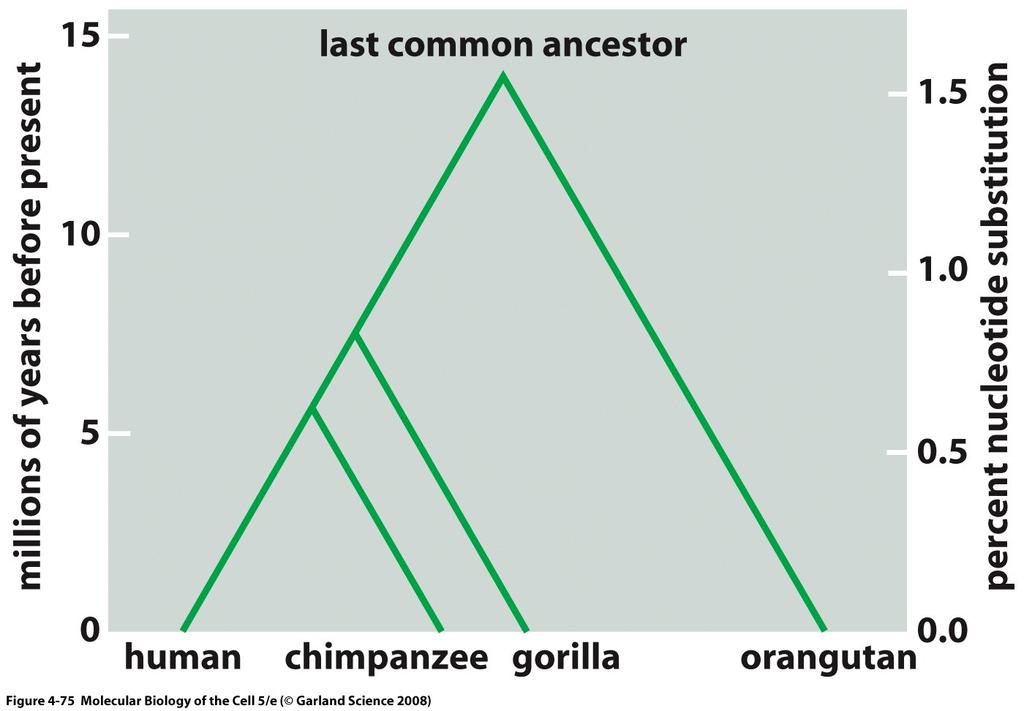
9 Evolutionary Relationships
10 Homology vs Similarity Similarity is a measure of the quality of alignment between two sequences. High similarity is evidence for homology. Homology is an inference from similarity. Similar sequences may correspond to orthologs or paralogs*. * Or, possibly, they derived from common selective pressures rather than a common ancestor. Or, the organisms were exposed to a common virus. Or,
11 Part 2: Sequence Alignment
12 Pairwise Sequence Alignment Sequence similarity depends on an alignment. What is an alignment, and why might it be significant? An alignment is a mapping from one sequence to another. Biological alignment maps together elements that are likely to have arisen from a common ancestor The existence of an alignment with many matches is an indication of homology
13 What complicates sequence alignment? Evolutionary changes Genetic variation Mutations (e.g. SNPs) Copy number variation Duplications, inversions, translocations, segment shuffling Insertions, Deletions, Substitutions
14 What counts as similarity? Similarity can be defined by counting positions that match between two sequences But which positions? Allowing gaps makes a difference in the number of matching positions abcdef abcdef abcdef- abceef acdefg a-cdefg
15 Not all mismatches are the same Some amino acids are more substitutable for each other than others. Serine and threonine are more alike than tryptophan and alanine. We can introduce "mismatch costs" for handling different substitutions. We don't usually use mismatch costs in aligning nucleotide sequences, since no substitution is per se better than any other.
16 Many possible alignments to consider Without gaps, there are are N+M-1 possible alignments between sequences of length N and M Once we start allowing gaps, there are many more possible arrangements to consider: abcbcd abcbcd abcbcd abc--d a--bcd ab--cd This becomes a very large number when we allow mismatches, since we then need to look at every possible pairing between elements: there are roughly N M possible alignments. Aligning length 100 sequences this way is impractical
17 Avoiding random alignments with a score function Not only are there many possible gapped alignments, but introducing too many gaps makes nonsense alignments possible: s--e-----qu---en--ce (sequence) sometimesquipsentice Want to distinguish between alignments that occur due to homology, and those that could be expected to be seen just by chance. Define a score function that accounts for both element mismatches and a gap penalty

18 Match scores are often calculated on the basis of the frequency of particular mutations in very similar sequences. We can transform substitution frequencies into log odds scores, which can then be added together. Match scores
19 An alignment score An alignment score is the sum of all the match scores of an alignment, with a penalty subtracted for each gap. Gap penalties are usually "affine" meaning that the penalty for one long gap is smaller than the penalty for many smaller gaps that add up to the same size. Match score Gap start + continuation penalty a b c - - d a c c e f d => 24 - (10 + 2) = 12 Alignment Score
20 Global & Local alignments A global alignment includes all elements of a sequence, and includes gaps A global alignment may or may not include "end gap" penalties. And.--so,.from.hour.to.hour,.we.ripe.and.ripe And.then,.from.hour.to.hour,.we.rot-.and.rot- A local alignment includes only subsequences, and sometimes is computed without gaps. My.care.is.loss.of.care,.by.old.care.done, Your.care.is.gain.of.care,.by.new.care.won
21 Local vs. Global alignments Local alignments can find shared domains in divergent proteins and are fast to compute Global alignments are better indicators of homology and take longer to compute.
22 Finding the optimal alignment Given a pair of sequences and a score function, identify the best scoring (optimal) alignment between the sequences. Remember, exponential number of possible alignments (most with terrible scores). Computer science to the rescue: dynamic programming identifies optimal alignments in time proportional to the sum of the lengths of the sequences
23 A brief aside on Computational Complexity A key idea in computer science: How much work does it take to solve a class of problems? How do we measure complexity? Relative to problem size How long does it take? Clock time versus operations Order: O(?) notation Worst case / best case Other resources used (particularly space)
24 Dynamic programming The key idea is to break the larger problem down into smaller sub-problems which are solved, the results stored, and then combined. DP is usually applied to optimization problems. Here, we start aligning the sequences left to right Once a prefix is optimally aligned, nothing about the remainder of the alignment can change the alignment of the prefix. We construct a matrix of possible alignment scores (NxM 2 calculations worst case) and then "traceback" to find the optimal alignment. Called Needleman-Wunsch or Smith-Waterman
25 Dynamic programming alignment Each cell contains the score for the best aligned sequence prefix up to that position. Start by filling in initial gap and first element to first element match score Use arrow to indicate path to that alignment Align ACD to AACADCD: (match = 5, gap start = -5, gap continue = -2)
26 Continue filling in optimal path scores For each cell, have three choices for how to get there from the last optimal alignment (match, gap sequence 1, gap sequence 2). Best score(s) are selected, and arrows added indicated route. From -5 align As = 0 From 5, insert gap = 0 From -7, insert gap = A -A AA A A A- AA -- AA --A AA- align As insert gap insert gap
27 Optimal alignment by traceback We traceback a path that gets us the highest score. If we don't have end gap penalties, then take any path from the last row or column to the first. Otherwise we need to include the top and bottom corners AACADCD AACADCD -AC-D-- ---A-CD
28 Parameter Selection The optimal alignment between a pair of sequences depends critically on the selection of the score matrix and the gap penalty. These sorts of generic inputs to a program are called parameters. How do we pick the ones that give the most biologically meaningful alignments (and alignment scores?)
29 How do we pick match scores? For match scores, two main options PAM based on global alignments of closely related sequences. Normalized to changes per 100 sites, then exponentiated for more distant relatives. BLOSUM based on local alignments in much more diverse sequences Each matrix has versions aimed at different evolutionary distances. BLOSUM62 is NCBI s default. BLOSUM45 may work better for more evolutionarily distant sequences.
30 Picking gap penalties Many different possible forms: Most common is affine (gap open + gap continue penalities) More complex penalties have been proposed. Penalties must be commensurate with match scores. Therefore, the match scoring scheme influences the gap penalty Most alignment programs suggest appropriate penalties for each match score option.
31 Searching for optimal scores One possibility is to try several different match score and gap penalties, and choose the best In general, this is called parameter space search and it is important in many areas. Problems requires a lot computation we need some principled way to compare the results. Use significance testing to compare...
32 The significance of an alignment Significance testing is the branch of statistics that is concerned with assessing the probability that a particular result could have occurred by chance. How do we calculate the probability that an alignment occurred by chance? Either with a model of evolution, or Empirically, by scrambling our sequences and calculating scores on many randomized (and by assumption unrelated) sequences. Incorporated into BLAST: E-value
33 Part 3: Search
34 Linear search Test query against each target sequentially Worst case, query matches last target and you have as many tests as targets (size of database) Query TTACG Average case, test half the targets. Linear in the size of the database Database ACTGA TTAGG CGTAA AGAGA CGATA CCGGA GCCCT TTACG
35 Indexed (binary) search Create a sorted set of keys that point to entries Start in the middle, then figure out which half Eliminate half the database each step, so need log 2 steps at worst Need to build the index (takes space and time at each database update) Query TTACG 1 2 Index ACTGA AGAGA CCGGA 3 CGATA CGTAA GCCCT TTACG TTAGG Database ACTGA TTAGG CGTAA AGAGA CGATA CCGGA GCCCT TTACG
36 Hash tables Map each query to an arbitrary number with a hash function Use those numbers as an index into a table Collisions can happen, but are rare Constant time lookup, no index construction f (TTACG)= 8 Hash table 1. CGATA 2. GCCCT 3. CGTAA, AGAGA ACTGA 6. CCGGA 7. TTAGG 8. TTACG
37 How to define a hash function Basic: must map keys to a number that is within the size of the table Desired: minimize collisions So: similar keys should lead to different hashes Good general method: map key to a number, and then take the remainder when divided by a prime number. Specialized hash functions can be better. Hash tables are the basis of most database lookups.
38 Approximate searches Recall the needs of sequence searches: Not looking for exact match, but similar sequences Database search methods only help us find exact matches. Hash tables particularly bad at similar because we need similar keys to map to different hashes First, need to define what is similar, then find efficient ways to search for similar sequences.
39 Part 4: BLAST Basic Local Alignment Search Tool
40 Why BLAST? Dynamic programming solutions to alignment problems are relatively slow, and don't lend themselves to efficient database search. Time complexity proportional to the size of the database. Need some way to search a large database to find sequences that have an inexact match to a query sequence BLAST: an imperfect approximation to DP. DP finds some distantly related sequences the approximations don't
41 Sequence search basics BLAST is x faster than DP Proper use is similar to DP: Use appropriate substitution and gap scores BLOSUM62 is good for weak protein similarities Use PAM30, PAM70 or BLOSUM45 for better results on more similar sequences, BLOSUM80 for most distant Use low-complexity (repetitive seq) filters and filter out human repeats (ALUs, etc) If searching for coding regions, always translate nucleotide to amino acid sequence.
42 How BLAST works Break sequence into overlapping words, by default of length 3. Sequence of length n makes n-m+1 m-size words ABCDE ABC, BCD, CDE For each word, define ~50 other words that are similar (use substitution matrix + threshold T) Repeat for each of the n-m+1 words, giving about 50*n words (out of 20 3 =8000 possible) Use a hash table to find all places in DB with an exact match to any of those words.
43 Extending HSPs Identify database sequences that contain several matching words on the same diagonal (think DP alignments) and within a short distance. Extend these short, ungapped alignments in both directions along the sequence so long as score of alignment increases. BLAST alignments scored simply with a log-odds matrix; no gap penalties at this point. Call these extended alignments HSPs for high scoring pairs
44 Is an HSP Significant? What is the probability of scoring at least as large as x by chance? Extreme value (not Normal!) distribution: Where m is size of the database, n is length of query, and l is average length of alignment between two random sequences of those lengths using this scoring scheme. Called E value for expectation (analogous to p value) High BLAST score = low E value (low probability of chance)
45 K and λ Parameters of the extreme value distribution Depend on the particular substitution matrix Estimated by aligning a lot of random sequences drawn on a particular distribution of amino acids, and fitting the extreme value distribution to those alignments These empirical estimates may not be correct (error in the assumed distribution of AAs used to create the random sequences) but seem to be reasonably close.
46 BLAST2: add gaps Multiple HSPs in one target sequence possibility of gapped alignment. Combine HSP scores to score whole sequence: Add HSP scores Adjust K and λ for this scoring method Set modest e-value threshold to identify reasonable target set Use DP to produce final gapped alignments Run DP on the (relatively) small number of database sequences that were above the threshold with multiple HSPs
47 Practical Gapped BLAST Default on NCBI web site BLAST versus DP on whole databases Still might miss some alignments DP would find as database search tool DP on fractions of the database (e.g. all human sequences) can be done with parallel hardware, but computational complexity scales with database size. BLAST allows users to set certain gap penalties, word sizes and thresholds in Advanced settings but not all (since K & λ have to be calculated in advance)
48 Part 5: Closing comments
49 Motivating scenarios "I have just sequenced a DNA fragment Run a BLAST search Once you have candidates, run a more careful alignment among them. "I've located a gene using a gene-finding algorithm Run BLAST to locate similar genes. Run a global alignment to see differences. "I'm confirming a sequencing experiment do a global alignment From:
50 Study guide... Dynamic programming alignments are a key technology in bioinformatics, and you should understand how they work. The method is perhaps counterintuitive Work some examples by hand. All of the textbooks describe D-P, and there is more detail and supplementary material on the course web site.
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Brief review from last class
 Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD
 Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
BGGN 213 Foundations of Bioinformatics Barry Grant
 BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
Pairwise Sequence Alignment. Zhongming Zhao, PhD
 Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships
 Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
Alignment of Pairs of Sequences
 Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
Introduction to BLAST with Protein Sequences. Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2
 Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Similarity Searches on Sequence Databases
 Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
B L A S T! BLAST: Basic local alignment search tool. Copyright notice. February 6, Pairwise alignment: key points. Outline of tonight s lecture
 February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
Pairwise Sequence Alignment: Dynamic Programming Algorithms. COMP Spring 2015 Luay Nakhleh, Rice University
 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
From Smith-Waterman to BLAST
 From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
Sequence comparison: Local alignment
 Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
Lecture 10. Sequence alignments
 Lecture 10 Sequence alignments Alignment algorithms: Overview Given a scoring system, we need to have an algorithm for finding an optimal alignment for a pair of sequences. We want to maximize the score
Lecture 10 Sequence alignments Alignment algorithms: Overview Given a scoring system, we need to have an algorithm for finding an optimal alignment for a pair of sequences. We want to maximize the score
Sequence alignment algorithms
 Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
Scoring and heuristic methods for sequence alignment CG 17
 Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Lecture 5 Advanced BLAST
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
BLAST - Basic Local Alignment Search Tool
 Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Data Mining Technologies for Bioinformatics Sequences
 Data Mining Technologies for Bioinformatics Sequences Deepak Garg Computer Science and Engineering Department Thapar Institute of Engineering & Tecnology, Patiala Abstract Main tool used for sequence alignment
Data Mining Technologies for Bioinformatics Sequences Deepak Garg Computer Science and Engineering Department Thapar Institute of Engineering & Tecnology, Patiala Abstract Main tool used for sequence alignment
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University
 1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
Algorithmic Approaches for Biological Data, Lecture #20
 Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:
 FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores.
 CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Bioinformatics explained: Smith-Waterman
 Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Introduction to Computational Molecular Biology
 18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
Today s Lecture. Edit graph & alignment algorithms. Local vs global Computational complexity of pairwise alignment Multiple sequence alignment
 Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
FastA & the chaining problem
 FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
A Design of a Hybrid System for DNA Sequence Alignment
 IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment
 Multiple Sequence Alignment") CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
Biochemistry 324 Bioinformatics. Multiple Sequence Alignment (MSA)
") Biochemistry 324 Bioinformatics Multiple Sequence Alignment (MSA) Big- Οh notation Greek omicron symbol Ο The Big-Oh notation indicates the complexity of an algorithm in terms of execution speed and storage
Biochemistry 324 Bioinformatics Multiple Sequence Alignment (MSA) Big- Οh notation Greek omicron symbol Ο The Big-Oh notation indicates the complexity of an algorithm in terms of execution speed and storage
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A
 BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A Steve Thompson: stthompson@valdosta.edu http://www.bioinfo4u.net 1 Similarity searching and homology First, just
BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A Steve Thompson: stthompson@valdosta.edu http://www.bioinfo4u.net 1 Similarity searching and homology First, just
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT
 OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
Dynamic Programming & Smith-Waterman algorithm
 m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
Sequence Alignment. part 2
 Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment Heuristics
 Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Distributed Protein Sequence Alignment
 Distributed Protein Sequence Alignment ABSTRACT J. Michael Meehan meehan@wwu.edu James Hearne hearne@wwu.edu Given the explosive growth of biological sequence databases and the computational complexity
Distributed Protein Sequence Alignment ABSTRACT J. Michael Meehan meehan@wwu.edu James Hearne hearne@wwu.edu Given the explosive growth of biological sequence databases and the computational complexity
PROTEIN MULTIPLE ALIGNMENT MOTIVATION: BACKGROUND: Marina Sirota
 Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
BLAST. Basic Local Alignment Search Tool. Used to quickly compare a protein or DNA sequence to a database.
 BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences
 BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
Searching Sequence Databases
 Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
Lecture 3: February Local Alignment: The Smith-Waterman Algorithm
 CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
Similarity searches in biological sequence databases
 Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Sequence Comparison: Dynamic Programming. Genome 373 Genomic Informatics Elhanan Borenstein
 Sequence omparison: Dynamic Programming Genome 373 Genomic Informatics Elhanan Borenstein quick review: hallenges Find the best global alignment of two sequences Find the best global alignment of multiple
Sequence omparison: Dynamic Programming Genome 373 Genomic Informatics Elhanan Borenstein quick review: hallenges Find the best global alignment of two sequences Find the best global alignment of multiple
CAP BLAST. BIOINFORMATICS Su-Shing Chen CISE. 8/20/2005 Su-Shing Chen, CISE 1
 CAP 5510-6 BLAST BIOINFORMATICS Su-Shing Chen CISE 8/20/2005 Su-Shing Chen, CISE 1 BLAST Basic Local Alignment Prof Search Su-Shing Chen Tool A Fast Pair-wise Alignment and Database Searching Tool 8/20/2005
CAP 5510-6 BLAST BIOINFORMATICS Su-Shing Chen CISE 8/20/2005 Su-Shing Chen, CISE 1 BLAST Basic Local Alignment Prof Search Su-Shing Chen Tool A Fast Pair-wise Alignment and Database Searching Tool 8/20/2005
Notes on Dynamic-Programming Sequence Alignment
 Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Database Similarity Searching
 An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
Central Issues in Biological Sequence Comparison
 Central Issues in Biological Sequence Comparison Definitions: What is one trying to find or optimize? Algorithms: Can one find the proposed object optimally or in reasonable time optimize? Statistics:
Central Issues in Biological Sequence Comparison Definitions: What is one trying to find or optimize? Algorithms: Can one find the proposed object optimally or in reasonable time optimize? Statistics:
ICB Fall G4120: Introduction to Computational Biology. Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology
 ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
Alignments BLAST, BLAT
 Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Important Example: Gene Sequence Matching. Corrigiendum. Central Dogma of Modern Biology. Genetics. How Nucleotides code for Amino Acids
 Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Chapter 4: Blast. Chaochun Wei Fall 2014
 Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Research Article Aligning Sequences by Minimum Description Length
 Hindawi Publishing Corporation EURASIP Journal on Bioinformatics and Systems Biology Volume 2007, Article ID 72936, 14 pages doi:10.1155/2007/72936 Research Article Aligning Sequences by Minimum Description
Hindawi Publishing Corporation EURASIP Journal on Bioinformatics and Systems Biology Volume 2007, Article ID 72936, 14 pages doi:10.1155/2007/72936 Research Article Aligning Sequences by Minimum Description
Lesson 13 Molecular Evolution
 Sequence Analysis Spring 2000 Dr. Richard Friedman (212)305-6901 (76901) friedman@cuccfa.ccc.columbia.edu 130BB Lesson 13 Molecular Evolution In this class we learn how to draw molecular evolutionary trees
Sequence Analysis Spring 2000 Dr. Richard Friedman (212)305-6901 (76901) friedman@cuccfa.ccc.columbia.edu 130BB Lesson 13 Molecular Evolution In this class we learn how to draw molecular evolutionary trees
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties
 Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
Comparative Analysis of Protein Alignment Algorithms in Parallel environment using CUDA
 Comparative Analysis of Protein Alignment Algorithms in Parallel environment using BLAST versus Smith-Waterman Shadman Fahim shadmanbracu09@gmail.com Shehabul Hossain rudrozzal@gmail.com Gulshan Jubaed
Comparative Analysis of Protein Alignment Algorithms in Parallel environment using BLAST versus Smith-Waterman Shadman Fahim shadmanbracu09@gmail.com Shehabul Hossain rudrozzal@gmail.com Gulshan Jubaed
Lecture 9: Core String Edits and Alignments
 Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Outline. Sequence Alignment. Types of Sequence Alignment. Genomics & Computational Biology. Section 2. How Computers Store Information
 enomics & omputational Biology Section Lan Zhang Sep. th, Outline How omputers Store Information Sequence lignment Dot Matrix nalysis Dynamic programming lobal: NeedlemanWunsch lgorithm Local: SmithWaterman
enomics & omputational Biology Section Lan Zhang Sep. th, Outline How omputers Store Information Sequence lignment Dot Matrix nalysis Dynamic programming lobal: NeedlemanWunsch lgorithm Local: SmithWaterman
Biological Sequence Matching Using Fuzzy Logic
 International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 1 Biological Sequence Matching Using Fuzzy Logic Nivit Gill, Shailendra Singh Abstract: Sequence alignment is the
International Journal of Scientific & Engineering Research Volume 2, Issue 7, July-2011 1 Biological Sequence Matching Using Fuzzy Logic Nivit Gill, Shailendra Singh Abstract: Sequence alignment is the
2) NCBI BLAST tutorial This is a users guide written by the education department at NCBI.
 NCBI BLAST tutorial This is a users guide written by the education department at NCBI.") Web resources -- Tour. page 1 of 8 This is a guided tour. Any homework is separate. In fact, this exercise is used for multiple classes and is publicly available to everyone. The entire tour will take
Web resources -- Tour. page 1 of 8 This is a guided tour. Any homework is separate. In fact, this exercise is used for multiple classes and is publicly available to everyone. The entire tour will take
Sequencing Alignment I
 Sequencing Alignment I Lectures 16 Nov 21, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline: Sequence
Sequencing Alignment I Lectures 16 Nov 21, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall (JHN) 022 1 Outline: Sequence
Bioinformatics 1: lecture 4. Followup of lecture 3? Molecular evolution Global, semi-global and local Affine gap penalty
 Bioinformatics 1: lecture 4 Followup of lecture 3? Molecular evolution Global, semi-global and local Affine gap penalty How sequences evolve point mutations (single base changes) deletion (loss of residues
Bioinformatics 1: lecture 4 Followup of lecture 3? Molecular evolution Global, semi-global and local Affine gap penalty How sequences evolve point mutations (single base changes) deletion (loss of residues
Multiple Sequence Alignment: Multidimensional. Biological Motivation
 Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Multiple Sequence Alignment: Multidimensional Dynamic Programming Boston University Biological Motivation Compare a new sequence with the sequences in a protein family. Proteins can be categorized into
Profiles and Multiple Alignments. COMP 571 Luay Nakhleh, Rice University
 Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
Profiles and Multiple Alignments COMP 571 Luay Nakhleh, Rice University Outline Profiles and sequence logos Profile hidden Markov models Aligning profiles Multiple sequence alignment by gradual sequence
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 13 G R A T I V. Iterative homology searching,
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use