Adam M Phillippy Center for Bioinformatics and Computational Biology
|
|
|
- Bennett Bond
- 5 years ago
- Views:
Transcription

1 Adam M Phillippy Center for Bioinformatics and Computational Biology
2 WGS sequencing shearing sequencing assembly
3 WGS assembly Overlap reads identify reads with shared k-mers calculate edit distance Layout reads walk the overlap graph hierarchically build contigs Generate consensus multi-align read layouts
4 Limitations of WGS Algorithmically hard Overlap reads 70,000 choose 2 = 2.5 billion combinations hard for large eukaryotic genomes Layout reads interpret the overlap graph hard for low coverage projects (too few edges) hard for repetitive projects (too many edges)
5 AMOScmp overview Pick a reference sequence assembly template Align target reads to the reference 2.5 billion 70,000 combinations Infer read relationships from alignments if their mappings overlap, they must overlap Create read layout fine tune the mappings Build a consensus
6 Picking a reference The closer the better sequence similarity high identity structural similarity similar repeat distributions few rearrangements Preferably complete non-contiguous reference fragmented results forced alignments singletons
7 Mapping the reads Generate read to reference alignments using MUMmer (nucmer) Pick the correct alignments using modified LIS algorithm allow fragmented mappings allow multiple, equivalent mappings Select repeat copies use mate information randomly place leftovers
8 Read alignments read reference
9 Problem Longest Increasing Subsequence For a list of n integers, find the longest strictly increasing subsequence from left to right Complexity O(n log n) via greedy set cover O(n 2 ) via dynamic programming O(l) for n < l / log l
10 LIS for alignments Alignments are not integers S i = S j + (len i * idy i ) max(olapr ij, olapq ij ) reward greater length and identity force mutually consistent ordering penalize overlap
11 LIS with repeats Problem For a list of n integers, find a set of disjoint subsequences within a given length of the LIS
12 Repeat selection
13 Making the layout Locate all alignment breaks For each break, count yay and nay reads scan across the reference from left to right read heap contains all the spanning reads count supporting, discounting, fuzzy keep the majority and toss the minority OR toss everything Adjust for polymorphism reads inside an insertion need to be handled separately reads after an insertion need to be offset accordingly Worst case O(cr log r)
14 Alignment breaks
15 Insertions Reference B A Reference A Target B A Target A Reference A B C Reference A B Target Insertion A B C Target A B Insertion
16 Rearrangement B Reference A C Target A B C C Reference I A III II B IV Target I II III IV A B C
17 Validating conflicts heap
18 Handling inserts R Q R Q
19 Example results Target Streptococcus agalactiae 2603 V/R Reference Streptococcus agalactiae NEM316 Streptococcus agalactiae 2603 V/R
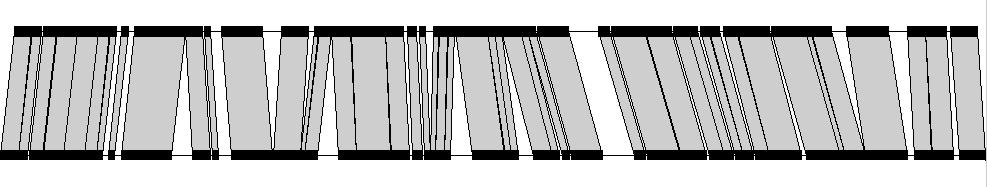
20
21 2603 read placement NEM 316 reference 29,456 alignments ~23,000 after LIS 26,099 total reads 21,816 unique 148 unique mate 22 mate constraints 443 random 3670 unplaced Self reference 34,846 alignments ~26,000 after LIS 26,099 total reads 25,301 unique 314 unique mate 22 mate constraints 442 random 20 unplaced
22 2603 read layout NEM 316 reference 312 conflicts 34 accepted 185 rejected 93 unknown 155 contigs Self reference 138 conflicts 0 accepted 133 rejected 5 unknown 86 contigs
23 2603 assembly X N vs 2603 vs. NEM 316 CelAsm total total total contig size N50 N contig size N50 N contig size vs 2603 vs NEM 316 CelAsm LW X gaps gap size coverage gaps gap size coverage gaps gap size coverage coverage ,168, ,329, ,261, , , , , , , , , , , , , , , , N ,001, , , ,593,364 2, ,393,287 1, ,488,287 1, ,856,394 5, ,640,231 4, ,812,266 4, ,043,842 14, ,829,976 10, ,046,730 12, ,100,541 27, ,891,527 18, ,110,396 21, ,119,579 42, ,919,237 24, ,132,490 33,953
24 Benefits Low coverage projects very thin overlaps permissible larger contigs higher assembly confidence High coverage projects algorithmically simplified fewer misassemblies given a good reference and implementation greatly reduced time and memory requirements under 5 min / 100 MB for a 5 Mbp genome more reads included in the assembly

25
26 Applications Low coverage projects thin overlaps make for bigger contigs allow for earlier SNP detection Environmental sequencing hybrid assembly of multiple strains Short read sequencing traditional algorithms fail for short reads overlaps too short, coverage too deep, non-uniform coverage Assembly validation self reference alignment breaks tandem collapse polymorphism
27 Open questions Hybrid assembly conventional / comparative who comes first? Read mapping repeats increase runtime sensitivity / specificity exact matches only Layout missing sequence inexact repeat copies identity cutoff surrogates polymorphisms query insertions assembly separately bambus rearrangements / tandems examine location
28 Mihai Pop, Adam Phillippy, Arthur L. Delcher,, Steven L. Salzberg. Comparative genome assembly. Briefings in Bioinformatics Sep; 5(3): Mihai Pop Arthur Delcher Steven Salzberg Stefan Kurtz NIH Michael Schatz Pawel Gajer
AMOS Assembly Validation and Visualization
 AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland April 7, 2006 Outline AMOS Introduction Getting Data into AMOS AMOS
AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland April 7, 2006 Outline AMOS Introduction Getting Data into AMOS AMOS
Description of a genome assembler: CABOG
 Theo Zimmermann Description of a genome assembler: CABOG CABOG (Celera Assembler with the Best Overlap Graph) is an assembler built upon the Celera Assembler, which, at first, was designed for Sanger sequencing,
Theo Zimmermann Description of a genome assembler: CABOG CABOG (Celera Assembler with the Best Overlap Graph) is an assembler built upon the Celera Assembler, which, at first, was designed for Sanger sequencing,
Constrained traversal of repeats with paired sequences
 RECOMB 2011 Satellite Workshop on Massively Parallel Sequencing (RECOMB-seq) 26-27 March 2011, Vancouver, BC, Canada; Short talk: 2011-03-27 12:10-12:30 (presentation: 15 minutes, questions: 5 minutes)
RECOMB 2011 Satellite Workshop on Massively Parallel Sequencing (RECOMB-seq) 26-27 March 2011, Vancouver, BC, Canada; Short talk: 2011-03-27 12:10-12:30 (presentation: 15 minutes, questions: 5 minutes)
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
Read Mapping. Slides by Carl Kingsford
 Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Reducing Genome Assembly Complexity with Optical Maps
 Reducing Genome Assembly Complexity with Optical Maps AMSC 663 Mid-Year Progress Report 12/13/2011 Lee Mendelowitz Lmendelo@math.umd.edu Advisor: Mihai Pop mpop@umiacs.umd.edu Computer Science Department
Reducing Genome Assembly Complexity with Optical Maps AMSC 663 Mid-Year Progress Report 12/13/2011 Lee Mendelowitz Lmendelo@math.umd.edu Advisor: Mihai Pop mpop@umiacs.umd.edu Computer Science Department
Preliminary Syllabus. Genomics. Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification
 Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Building approximate overlap graphs for DNA assembly using random-permutations-based search.
 An algorithm is presented for fast construction of graphs of reads, where an edge between two reads indicates an approximate overlap between the reads. Since the algorithm finds approximate overlaps directly,
An algorithm is presented for fast construction of graphs of reads, where an edge between two reads indicates an approximate overlap between the reads. Since the algorithm finds approximate overlaps directly,
Reducing Genome Assembly Complexity with Optical Maps
 Reducing Genome Assembly Complexity with Optical Maps Lee Mendelowitz LMendelo@math.umd.edu Advisor: Dr. Mihai Pop Computer Science Department Center for Bioinformatics and Computational Biology mpop@umiacs.umd.edu
Reducing Genome Assembly Complexity with Optical Maps Lee Mendelowitz LMendelo@math.umd.edu Advisor: Dr. Mihai Pop Computer Science Department Center for Bioinformatics and Computational Biology mpop@umiacs.umd.edu
Scalable Solutions for DNA Sequence Analysis
 Scalable Solutions for DNA Sequence Analysis Michael Schatz Dec 4, 2009 JHU/UMD Joint Sequencing Meeting The Evolution of DNA Sequencing Year Genome Technology Cost 2001 Venter et al. Sanger (ABI) $300,000,000
Scalable Solutions for DNA Sequence Analysis Michael Schatz Dec 4, 2009 JHU/UMD Joint Sequencing Meeting The Evolution of DNA Sequencing Year Genome Technology Cost 2001 Venter et al. Sanger (ABI) $300,000,000
(for more info see:
 Genome assembly (for more info see: http://www.cbcb.umd.edu/research/assembly_primer.shtml) Introduction Sequencing technologies can only "read" short fragments from a genome. Reconstructing the entire
Genome assembly (for more info see: http://www.cbcb.umd.edu/research/assembly_primer.shtml) Introduction Sequencing technologies can only "read" short fragments from a genome. Reconstructing the entire
Genome 373: Genome Assembly. Doug Fowler
 Genome 373: Genome Assembly Doug Fowler What are some of the things we ve seen we can do with HTS data? We ve seen that HTS can enable a wide variety of analyses ranging from ID ing variants to genome-
Genome 373: Genome Assembly Doug Fowler What are some of the things we ve seen we can do with HTS data? We ve seen that HTS can enable a wide variety of analyses ranging from ID ing variants to genome-
High-throughput Sequence Alignment using Graphics Processing Units
 High-throughput Sequence Alignment using Graphics Processing Units Michael Schatz & Cole Trapnell May 21, 2009 UMD NVIDIA CUDA Center Of Excellence Presentation Searching Wikipedia How do you find all
High-throughput Sequence Alignment using Graphics Processing Units Michael Schatz & Cole Trapnell May 21, 2009 UMD NVIDIA CUDA Center Of Excellence Presentation Searching Wikipedia How do you find all
Genome Assembly Using de Bruijn Graphs. Biostatistics 666
 Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Finding the appropriate method, with a special focus on: Mapping and alignment. Philip Clausen
 Finding the appropriate method, with a special focus on: Mapping and alignment Philip Clausen Background Most people choose their methods based on popularity and history, not by reasoning and research.
Finding the appropriate method, with a special focus on: Mapping and alignment Philip Clausen Background Most people choose their methods based on popularity and history, not by reasoning and research.
Reducing Genome Assembly Complexity with Optical Maps Mid-year Progress Report
 Reducing Genome Assembly Complexity with Optical Maps Mid-year Progress Report Lee Mendelowitz LMendelo@math.umd.edu Advisor: Dr. Mihai Pop Computer Science Department Center for Bioinformatics and Computational
Reducing Genome Assembly Complexity with Optical Maps Mid-year Progress Report Lee Mendelowitz LMendelo@math.umd.edu Advisor: Dr. Mihai Pop Computer Science Department Center for Bioinformatics and Computational
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS
 ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS Ivan Vogel Doctoral Degree Programme (1), FIT BUT E-mail: xvogel01@stud.fit.vutbr.cz Supervised by: Jaroslav Zendulka E-mail: zendulka@fit.vutbr.cz
ON HEURISTIC METHODS IN NEXT-GENERATION SEQUENCING DATA ANALYSIS Ivan Vogel Doctoral Degree Programme (1), FIT BUT E-mail: xvogel01@stud.fit.vutbr.cz Supervised by: Jaroslav Zendulka E-mail: zendulka@fit.vutbr.cz
Confidence Intervals. Dennis Sun Data 301
 Dennis Sun Data 301 Statistical Inference probability Population / Box Sample / Data statistics The goal of statistics is to infer the unknown population from the sample. We ve already seen one mode of
Dennis Sun Data 301 Statistical Inference probability Population / Box Sample / Data statistics The goal of statistics is to infer the unknown population from the sample. We ve already seen one mode of
Genome 373: Mapping Short Sequence Reads I. Doug Fowler
 Genome 373: Mapping Short Sequence Reads I Doug Fowler Two different strategies for parallel amplification BRIDGE PCR EMULSION PCR Two different strategies for parallel amplification BRIDGE PCR EMULSION
Genome 373: Mapping Short Sequence Reads I Doug Fowler Two different strategies for parallel amplification BRIDGE PCR EMULSION PCR Two different strategies for parallel amplification BRIDGE PCR EMULSION
Read Mapping and Assembly
 Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Statistical Bioinformatics: Read Mapping and Assembly Stefan Seemann seemann@rth.dk University of Copenhagen April 9th 2019 Why sequencing? Why sequencing? Which organism does the sample comes from? Assembling
Sequence Assembly. BMI/CS 576 Mark Craven Some sequencing successes
 Sequence Assembly BMI/CS 576 www.biostat.wisc.edu/bmi576/ Mark Craven craven@biostat.wisc.edu Some sequencing successes Yersinia pestis Cannabis sativa The sequencing problem We want to determine the identity
Sequence Assembly BMI/CS 576 www.biostat.wisc.edu/bmi576/ Mark Craven craven@biostat.wisc.edu Some sequencing successes Yersinia pestis Cannabis sativa The sequencing problem We want to determine the identity
IDBA - A practical Iterative de Bruijn Graph De Novo Assembler
 IDBA - A practical Iterative de Bruijn Graph De Novo Assembler Speaker: Gabriele Capannini May 21, 2010 Introduction De Novo Assembly assembling reads together so that they form a new, previously unknown
IDBA - A practical Iterative de Bruijn Graph De Novo Assembler Speaker: Gabriele Capannini May 21, 2010 Introduction De Novo Assembly assembling reads together so that they form a new, previously unknown
Introduction to Genome Assembly. Tandy Warnow
 Introduction to Genome Assembly Tandy Warnow 2 Shotgun DNA Sequencing DNA target sample SHEAR & SIZE End Reads / Mate Pairs 550bp 10,000bp Not all sequencing technologies produce mate-pairs. Different
Introduction to Genome Assembly Tandy Warnow 2 Shotgun DNA Sequencing DNA target sample SHEAR & SIZE End Reads / Mate Pairs 550bp 10,000bp Not all sequencing technologies produce mate-pairs. Different
I519 Introduction to Bioinformatics, Genome assembly. Yuzhen Ye School of Informatics & Computing, IUB
 I519 Introduction to Bioinformatics, 2014 Genome assembly Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Contents Genome assembly problem Approaches Comparative assembly The string
I519 Introduction to Bioinformatics, 2014 Genome assembly Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Contents Genome assembly problem Approaches Comparative assembly The string
Sequence Assembly Required!
 Sequence Assembly Required! 1 October 3, ISMB 20172007 1 Sequence Assembly Genome Sequenced Fragments (reads) Assembled Contigs Finished Genome 2 Greedy solution is bounded 3 Typical assembly strategy
Sequence Assembly Required! 1 October 3, ISMB 20172007 1 Sequence Assembly Genome Sequenced Fragments (reads) Assembled Contigs Finished Genome 2 Greedy solution is bounded 3 Typical assembly strategy
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
1 Abstract. 2 Introduction. 3 Requirements
 1 Abstract 2 Introduction This SOP describes the HMP Whole- Metagenome Annotation Pipeline run at CBCB. This pipeline generates a 'Pretty Good Assembly' - a reasonable attempt at reconstructing pieces
1 Abstract 2 Introduction This SOP describes the HMP Whole- Metagenome Annotation Pipeline run at CBCB. This pipeline generates a 'Pretty Good Assembly' - a reasonable attempt at reconstructing pieces
Running SNAP. The SNAP Team October 2012
 Running SNAP The SNAP Team October 2012 1 Introduction SNAP is a tool that is intended to serve as the read aligner in a gene sequencing pipeline. Its theory of operation is described in Faster and More
Running SNAP The SNAP Team October 2012 1 Introduction SNAP is a tool that is intended to serve as the read aligner in a gene sequencing pipeline. Its theory of operation is described in Faster and More
Finishing Circular Assemblies. J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015
 Finishing Circular Assemblies J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Assembly Strategies de Bruijn graph Velvet, ABySS earlier, basic assemblers IDBA, SPAdes later, multi-k
Finishing Circular Assemblies J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Assembly Strategies de Bruijn graph Velvet, ABySS earlier, basic assemblers IDBA, SPAdes later, multi-k
Genome Assembly and De Novo RNAseq
 Genome Assembly and De Novo RNAseq BMI 7830 Kun Huang Department of Biomedical Informatics The Ohio State University Outline Problem formulation Hamiltonian path formulation Euler path and de Bruijin graph
Genome Assembly and De Novo RNAseq BMI 7830 Kun Huang Department of Biomedical Informatics The Ohio State University Outline Problem formulation Hamiltonian path formulation Euler path and de Bruijin graph
QB LECTURE #1: Algorithms and Dynamic Programming
 QB LECTURE #1: Algorithms and Dynamic Programming Adam Siepel Nov. 16, 2015 2 Plan for Today Introduction to algorithms Simple algorithms and running time Dynamic programming Soon: sequence alignment 3
QB LECTURE #1: Algorithms and Dynamic Programming Adam Siepel Nov. 16, 2015 2 Plan for Today Introduction to algorithms Simple algorithms and running time Dynamic programming Soon: sequence alignment 3
Kraken: ultrafast metagenomic sequence classification using exact alignments
 Kraken: ultrafast metagenomic sequence classification using exact alignments Derrick E. Wood and Steven L. Salzberg Bioinformatics journal club October 8, 2014 Märt Roosaare Need for speed Metagenomic
Kraken: ultrafast metagenomic sequence classification using exact alignments Derrick E. Wood and Steven L. Salzberg Bioinformatics journal club October 8, 2014 Märt Roosaare Need for speed Metagenomic
Tour Guide for Windows and Macintosh
 Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
splitmem: graphical pan-genome analysis with suffix skips Shoshana Marcus May 7, 2014
 splitmem: graphical pan-genome analysis with suffix skips Shoshana Marcus May 7, 2014 Outline 1 Overview 2 Data Structures 3 splitmem Algorithm 4 Pan-genome Analysis Objective Input! Output! A B C D Several
splitmem: graphical pan-genome analysis with suffix skips Shoshana Marcus May 7, 2014 Outline 1 Overview 2 Data Structures 3 splitmem Algorithm 4 Pan-genome Analysis Objective Input! Output! A B C D Several
Pick a time window size w. In time span w, are there, Multiple References, to nearby addresses: Spatial Locality
 Pick a time window size w. In time span w, are there, Multiple References, to nearby addresses: Spatial Locality Repeated References, to a set of locations: Temporal Locality Take advantage of behavior
Pick a time window size w. In time span w, are there, Multiple References, to nearby addresses: Spatial Locality Repeated References, to a set of locations: Temporal Locality Take advantage of behavior
Omega: an Overlap-graph de novo Assembler for Metagenomics
 Omega: an Overlap-graph de novo Assembler for Metagenomics B a h l e l H a i d e r, Ta e - H y u k A h n, B r i a n B u s h n e l l, J u a n j u a n C h a i, A l e x C o p e l a n d, C h o n g l e Pa n
Omega: an Overlap-graph de novo Assembler for Metagenomics B a h l e l H a i d e r, Ta e - H y u k A h n, B r i a n B u s h n e l l, J u a n j u a n C h a i, A l e x C o p e l a n d, C h o n g l e Pa n
Reevaluating Assembly Evaluations using Feature Analysis: GAGE and Assemblathons Supplementary material
 Reevaluating Assembly Evaluations using Feature Analysis: GAGE and Assemblathons Supplementary material Francesco Vezzi, Giuseppe Narzisi, Bud Mishra September 29, 22 Features computation FRC bam computes
Reevaluating Assembly Evaluations using Feature Analysis: GAGE and Assemblathons Supplementary material Francesco Vezzi, Giuseppe Narzisi, Bud Mishra September 29, 22 Features computation FRC bam computes
Tutorial. Aligning contigs manually using the Genome Finishing. Sample to Insight. February 6, 2019
 Aligning contigs manually using the Genome Finishing Module February 6, 2019 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Aligning contigs manually using the Genome Finishing Module February 6, 2019 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Generating a Genome Assembly with PCAP
 Generating a Genome Assembly with UNIT 11.3 In recent years, the whole-genome shotgun (WGS) technique has become the method of choice for generating genome sequences. In this technique, the entire genome
Generating a Genome Assembly with UNIT 11.3 In recent years, the whole-genome shotgun (WGS) technique has become the method of choice for generating genome sequences. In this technique, the entire genome
Shotgun sequencing. Coverage is simply the average number of reads that overlap each true base in genome.
 Shotgun sequencing Genome (unknown) Reads (randomly chosen; have errors) Coverage is simply the average number of reads that overlap each true base in genome. Here, the coverage is ~10 just draw a line
Shotgun sequencing Genome (unknown) Reads (randomly chosen; have errors) Coverage is simply the average number of reads that overlap each true base in genome. Here, the coverage is ~10 just draw a line
Computational models for bionformatics
 Computational models for bionformatics De-novo assembly and alignment-free measures Michele Schimd Department of Information Engineering July 8th, 2015 Michele Schimd (DEI) PostDoc @ DEI July 8th, 2015
Computational models for bionformatics De-novo assembly and alignment-free measures Michele Schimd Department of Information Engineering July 8th, 2015 Michele Schimd (DEI) PostDoc @ DEI July 8th, 2015
Towards a de novo short read assembler for large genomes using cloud computing
 Towards a de novo short read assembler for large genomes using cloud computing Michael Schatz April 21, 2009 AMSC664 Advanced Scientific Computing Outline 1.! Genome assembly by analogy 2.! DNA sequencing
Towards a de novo short read assembler for large genomes using cloud computing Michael Schatz April 21, 2009 AMSC664 Advanced Scientific Computing Outline 1.! Genome assembly by analogy 2.! DNA sequencing
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Space Efficient Linear Time Construction of
 Space Efficient Linear Time Construction of Suffix Arrays Pang Ko and Srinivas Aluru Dept. of Electrical and Computer Engineering 1 Laurence H. Baker Center for Bioinformatics and Biological Statistics
Space Efficient Linear Time Construction of Suffix Arrays Pang Ko and Srinivas Aluru Dept. of Electrical and Computer Engineering 1 Laurence H. Baker Center for Bioinformatics and Biological Statistics
MacVector for Mac OS X. The online updater for this release is MB in size
 MacVector 17.0.3 for Mac OS X The online updater for this release is 143.5 MB in size You must be running MacVector 15.5.4 or later for this updater to work! System Requirements MacVector 17.0 is supported
MacVector 17.0.3 for Mac OS X The online updater for this release is 143.5 MB in size You must be running MacVector 15.5.4 or later for this updater to work! System Requirements MacVector 17.0 is supported
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units
 GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
Fuzzy Classification of Genome Sequences Prior to Assembly Based on Similarity Measures *
 Fuzzy Classification of Genome Sequences Prior to Assembly Based on Similarity Measures * Sara Nasser, Gregory L. Vert, Adrienne Breland and Monica Nicolescu Department of Computer Science Engineering
Fuzzy Classification of Genome Sequences Prior to Assembly Based on Similarity Measures * Sara Nasser, Gregory L. Vert, Adrienne Breland and Monica Nicolescu Department of Computer Science Engineering
Assembly in the Clouds
 Assembly in the Clouds Michael Schatz October 13, 2010 Beyond the Genome Shredded Book Reconstruction Dickens accidentally shreds the first printing of A Tale of Two Cities Text printed on 5 long spools
Assembly in the Clouds Michael Schatz October 13, 2010 Beyond the Genome Shredded Book Reconstruction Dickens accidentally shreds the first printing of A Tale of Two Cities Text printed on 5 long spools
IDBA A Practical Iterative de Bruijn Graph De Novo Assembler
 IDBA A Practical Iterative de Bruijn Graph De Novo Assembler Yu Peng, Henry C.M. Leung, S.M. Yiu, and Francis Y.L. Chin Department of Computer Science, The University of Hong Kong Pokfulam Road, Hong Kong
IDBA A Practical Iterative de Bruijn Graph De Novo Assembler Yu Peng, Henry C.M. Leung, S.M. Yiu, and Francis Y.L. Chin Department of Computer Science, The University of Hong Kong Pokfulam Road, Hong Kong
De Novo Draft Genome Assembly Using Fuzzy K-mers
 De Novo Draft Genome Assembly Using Fuzzy K-mers John Healy Department of Computing & Mathematics Galway-Mayo Institute of Technology Ireland e-mail: john.healy@gmit.ie Abstract- Although second generation
De Novo Draft Genome Assembly Using Fuzzy K-mers John Healy Department of Computing & Mathematics Galway-Mayo Institute of Technology Ireland e-mail: john.healy@gmit.ie Abstract- Although second generation
Aligners. J Fass 21 June 2017
 Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Sanger Data Assembly in SeqMan Pro
 Sanger Data Assembly in SeqMan Pro DNASTAR provides two applications for assembling DNA sequence fragments: SeqMan NGen and SeqMan Pro. SeqMan NGen is primarily used to assemble Next Generation Sequencing
Sanger Data Assembly in SeqMan Pro DNASTAR provides two applications for assembling DNA sequence fragments: SeqMan NGen and SeqMan Pro. SeqMan NGen is primarily used to assemble Next Generation Sequencing
Computational Architecture of Cloud Environments Michael Schatz. April 1, 2010 NHGRI Cloud Computing Workshop
 Computational Architecture of Cloud Environments Michael Schatz April 1, 2010 NHGRI Cloud Computing Workshop Cloud Architecture Computation Input Output Nebulous question: Cloud computing = Utility computing
Computational Architecture of Cloud Environments Michael Schatz April 1, 2010 NHGRI Cloud Computing Workshop Cloud Architecture Computation Input Output Nebulous question: Cloud computing = Utility computing
C++ for System Developers with Design Pattern
 C++ for System Developers with Design Pattern Introduction: This course introduces the C++ language for use on real time and embedded applications. The first part of the course focuses on the language
C++ for System Developers with Design Pattern Introduction: This course introduces the C++ language for use on real time and embedded applications. The first part of the course focuses on the language
Running SNAP. The SNAP Team February 2012
 Running SNAP The SNAP Team February 2012 1 Introduction SNAP is a tool that is intended to serve as the read aligner in a gene sequencing pipeline. Its theory of operation is described in Faster and More
Running SNAP The SNAP Team February 2012 1 Introduction SNAP is a tool that is intended to serve as the read aligner in a gene sequencing pipeline. Its theory of operation is described in Faster and More
De novo genome assembly
 BioNumerics Tutorial: De novo genome assembly 1 Aims This tutorial describes a de novo assembly of a Staphylococcus aureus genome, using single-end and pairedend reads generated by an Illumina R Genome
BioNumerics Tutorial: De novo genome assembly 1 Aims This tutorial describes a de novo assembly of a Staphylococcus aureus genome, using single-end and pairedend reads generated by an Illumina R Genome
Dynamic Programming. Lecture Overview Introduction
 Lecture 12 Dynamic Programming 12.1 Overview Dynamic Programming is a powerful technique that allows one to solve many different types of problems in time O(n 2 ) or O(n 3 ) for which a naive approach
Lecture 12 Dynamic Programming 12.1 Overview Dynamic Programming is a powerful technique that allows one to solve many different types of problems in time O(n 2 ) or O(n 3 ) for which a naive approach
IDBA - A Practical Iterative de Bruijn Graph De Novo Assembler
 IDBA - A Practical Iterative de Bruijn Graph De Novo Assembler Yu Peng, Henry Leung, S.M. Yiu, Francis Y.L. Chin Department of Computer Science, The University of Hong Kong Pokfulam Road, Hong Kong {ypeng,
IDBA - A Practical Iterative de Bruijn Graph De Novo Assembler Yu Peng, Henry Leung, S.M. Yiu, Francis Y.L. Chin Department of Computer Science, The University of Hong Kong Pokfulam Road, Hong Kong {ypeng,
Read Mapping. de Novo Assembly. Genomics: Lecture #2 WS 2014/2015
 Mapping de Novo Assembly Institut für Medizinische Genetik und Humangenetik Charité Universitätsmedizin Berlin Genomics: Lecture #2 WS 2014/2015 Today Genome assembly: the basics Hamiltonian and Eulerian
Mapping de Novo Assembly Institut für Medizinische Genetik und Humangenetik Charité Universitätsmedizin Berlin Genomics: Lecture #2 WS 2014/2015 Today Genome assembly: the basics Hamiltonian and Eulerian
CS681: Advanced Topics in Computational Biology
 CS681: Advanced Topics in Computational Biology Can Alkan EA224 calkan@cs.bilkent.edu.tr Week 7 Lectures 2-3 http://www.cs.bilkent.edu.tr/~calkan/teaching/cs681/ Genome Assembly Test genome Random shearing
CS681: Advanced Topics in Computational Biology Can Alkan EA224 calkan@cs.bilkent.edu.tr Week 7 Lectures 2-3 http://www.cs.bilkent.edu.tr/~calkan/teaching/cs681/ Genome Assembly Test genome Random shearing
Performing whole genome SNP analysis with mapping performed locally
 BioNumerics Tutorial: Performing whole genome SNP analysis with mapping performed locally 1 Introduction 1.1 An introduction to whole genome SNP analysis A Single Nucleotide Polymorphism (SNP) is a variation
BioNumerics Tutorial: Performing whole genome SNP analysis with mapping performed locally 1 Introduction 1.1 An introduction to whole genome SNP analysis A Single Nucleotide Polymorphism (SNP) is a variation
CS 173, Lecture B Introduction to Genome Assembly (using Eulerian Graphs) Tandy Warnow
 Tandy Warnow") CS 173, Lecture B Introduction to Genome Assembly (using Eulerian Graphs) Tandy Warnow 2 Shotgun DNA Sequencing DNA target sample SHEAR & SIZE End Reads / Mate Pairs 550bp 10,000bp Not all sequencing technologies
CS 173, Lecture B Introduction to Genome Assembly (using Eulerian Graphs) Tandy Warnow 2 Shotgun DNA Sequencing DNA target sample SHEAR & SIZE End Reads / Mate Pairs 550bp 10,000bp Not all sequencing technologies
Supplementary Material for The Generalized PatchMatch Correspondence Algorithm
 Supplementary Material for The Generalized PatchMatch Correspondence Algorithm Connelly Barnes 1, Eli Shechtman 2, Dan B Goldman 2, Adam Finkelstein 1 1 Princeton University, 2 Adobe Systems 1 Overview
Supplementary Material for The Generalized PatchMatch Correspondence Algorithm Connelly Barnes 1, Eli Shechtman 2, Dan B Goldman 2, Adam Finkelstein 1 1 Princeton University, 2 Adobe Systems 1 Overview
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014
 Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014 Deciphering the information contained in DNA sequences began decades ago since the time of Sanger sequencing.
Review of Recent NGS Short Reads Alignment Tools BMI-231 final project, Chenxi Chen Spring 2014 Deciphering the information contained in DNA sequences began decades ago since the time of Sanger sequencing.
Page 1 of 20. ABySS-Explorer v1.3.0: User Manual
 Page 1 of 20 ABySS-Explorer v1.3.0: User Manual prepared by: Ka Ming Nip, Cydney Nielsen, Shaun Jackman, Inanc Birol Canada's Michael Smith Genome Sciences Centre November 2011 ABySS-Explorer is an interactive
Page 1 of 20 ABySS-Explorer v1.3.0: User Manual prepared by: Ka Ming Nip, Cydney Nielsen, Shaun Jackman, Inanc Birol Canada's Michael Smith Genome Sciences Centre November 2011 ABySS-Explorer is an interactive
Sequencing. Computational Biology IST Ana Teresa Freitas 2011/2012. (BACs) Whole-genome shotgun sequencing Celera Genomics
 Whole-genome shotgun sequencing Celera Genomics") Computational Biology IST Ana Teresa Freitas 2011/2012 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics (BACs) 1 Must take the fragments
Computational Biology IST Ana Teresa Freitas 2011/2012 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics (BACs) 1 Must take the fragments
Introduction and tutorial for SOAPdenovo. Xiaodong Fang Department of Science and BGI May, 2012
 Introduction and tutorial for SOAPdenovo Xiaodong Fang fangxd@genomics.org.cn Department of Science and Technology @ BGI May, 2012 Why de novo assembly? Genome is the genetic basis for different phenotypes
Introduction and tutorial for SOAPdenovo Xiaodong Fang fangxd@genomics.org.cn Department of Science and Technology @ BGI May, 2012 Why de novo assembly? Genome is the genetic basis for different phenotypes
BIOINFORMATICS. Integrating genome assemblies with MAIA
 BIOINFORMATICS Vol. 26 ECCB 2010, pages i433 i439 doi:10.1093/bioinformatics/btq366 Integrating genome assemblies with MAIA Jurgen Nijkamp 1,2,3,, Wynand Winterbach 1,4, Marcel van den Broek 2,3, Jean-Marc
BIOINFORMATICS Vol. 26 ECCB 2010, pages i433 i439 doi:10.1093/bioinformatics/btq366 Integrating genome assemblies with MAIA Jurgen Nijkamp 1,2,3,, Wynand Winterbach 1,4, Marcel van den Broek 2,3, Jean-Marc
RESEARCH TOPIC IN BIOINFORMANTIC
 RESEARCH TOPIC IN BIOINFORMANTIC GENOME ASSEMBLY Instructor: Dr. Yufeng Wu Noted by: February 25, 2012 Genome Assembly is a kind of string sequencing problems. As we all know, the human genome is very
RESEARCH TOPIC IN BIOINFORMANTIC GENOME ASSEMBLY Instructor: Dr. Yufeng Wu Noted by: February 25, 2012 Genome Assembly is a kind of string sequencing problems. As we all know, the human genome is very
Working with AppleScript
 Tutorial for Macintosh Working with AppleScript 2017 Gene Codes Corporation Gene Codes Corporation 525 Avis Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) +1.734.769.7249 (elsewhere) +1.734.769.7074
Tutorial for Macintosh Working with AppleScript 2017 Gene Codes Corporation Gene Codes Corporation 525 Avis Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) +1.734.769.7249 (elsewhere) +1.734.769.7074
Introduction to Computational Molecular Biology
 18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
DNA Fragment Assembly
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri DNA Fragment Assembly Overlap
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri DNA Fragment Assembly Overlap
A Suffix Tree Construction Algorithm for DNA Sequences
 A Suffix Tree Construction Algorithm for DNA Sequences Hongwei Huo School of Computer Science and Technol Xidian University Xi 'an 710071, China Vojislav Stojkovic Computer Science Department Morgan State
A Suffix Tree Construction Algorithm for DNA Sequences Hongwei Huo School of Computer Science and Technol Xidian University Xi 'an 710071, China Vojislav Stojkovic Computer Science Department Morgan State
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
A Genome Assembly Algorithm Designed for Single-Cell Sequencing
 SPAdes A Genome Assembly Algorithm Designed for Single-Cell Sequencing Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput
SPAdes A Genome Assembly Algorithm Designed for Single-Cell Sequencing Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput
TRELLIS+: AN EFFECTIVE APPROACH FOR INDEXING GENOME-SCALE SEQUENCES USING SUFFIX TREES
 TRELLIS+: AN EFFECTIVE APPROACH FOR INDEXING GENOME-SCALE SEQUENCES USING SUFFIX TREES BENJARATH PHOOPHAKDEE AND MOHAMMED J. ZAKI Dept. of Computer Science, Rensselaer Polytechnic Institute, Troy, NY,
TRELLIS+: AN EFFECTIVE APPROACH FOR INDEXING GENOME-SCALE SEQUENCES USING SUFFIX TREES BENJARATH PHOOPHAKDEE AND MOHAMMED J. ZAKI Dept. of Computer Science, Rensselaer Polytechnic Institute, Troy, NY,
CloudBurst: Highly Sensitive Read Mapping with MapReduce
 Bioinformatics Advance Access published April 8, 2009 Sequence Analysis CloudBurst: Highly Sensitive Read Mapping with MapReduce Michael C. Schatz* Center for Bioinformatics and Computational Biology,
Bioinformatics Advance Access published April 8, 2009 Sequence Analysis CloudBurst: Highly Sensitive Read Mapping with MapReduce Michael C. Schatz* Center for Bioinformatics and Computational Biology,
ABySS. Assembly By Short Sequences
 ABySS Assembly By Short Sequences ABySS Developed at Canada s Michael Smith Genome Sciences Centre Developed in response to memory demands of conventional DBG assembly methods Parallelizability Illumina
ABySS Assembly By Short Sequences ABySS Developed at Canada s Michael Smith Genome Sciences Centre Developed in response to memory demands of conventional DBG assembly methods Parallelizability Illumina
Title:- Instructions to run GS Assembler and Mapper Course # BIOL 8803 Special Topic on Computational Genomics Assembly Group
 Title:- Instructions to run GS Assembler and Mapper Course # BIOL 8803 Special Topic on Computational Genomics Assembly Group Contents 1. Genome Assembly... 3 1.0. Data and Projects... 3 1.1. GS De Novo
Title:- Instructions to run GS Assembler and Mapper Course # BIOL 8803 Special Topic on Computational Genomics Assembly Group Contents 1. Genome Assembly... 3 1.0. Data and Projects... 3 1.1. GS De Novo
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Fast Hierarchical Clustering via Dynamic Closest Pairs
 Fast Hierarchical Clustering via Dynamic Closest Pairs David Eppstein Dept. Information and Computer Science Univ. of California, Irvine http://www.ics.uci.edu/ eppstein/ 1 My Interest In Clustering What
Fast Hierarchical Clustering via Dynamic Closest Pairs David Eppstein Dept. Information and Computer Science Univ. of California, Irvine http://www.ics.uci.edu/ eppstein/ 1 My Interest In Clustering What
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
A THEORETICAL ANALYSIS OF SCALABILITY OF THE PARALLEL GENOME ASSEMBLY ALGORITHMS
 A THEORETICAL ANALYSIS OF SCALABILITY OF THE PARALLEL GENOME ASSEMBLY ALGORITHMS Munib Ahmed, Ishfaq Ahmad Department of Computer Science and Engineering, University of Texas At Arlington, Arlington, Texas
A THEORETICAL ANALYSIS OF SCALABILITY OF THE PARALLEL GENOME ASSEMBLY ALGORITHMS Munib Ahmed, Ishfaq Ahmad Department of Computer Science and Engineering, University of Texas At Arlington, Arlington, Texas
Tutorial: De Novo Assembly of Paired Data
 : De Novo Assembly of Paired Data September 20, 2013 CLC bio Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com support@clcbio.com : De Novo Assembly
: De Novo Assembly of Paired Data September 20, 2013 CLC bio Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com support@clcbio.com : De Novo Assembly
AMemoryEfficient Short Read De Novo Assembly Algorithm
 Original Paper AMemoryEfficient Short Read De Novo Assembly Algorithm Yuki Endo 1,a) Fubito Toyama 1 Chikafumi Chiba 2 Hiroshi Mori 1 Kenji Shoji 1 Received: October 17, 2014, Accepted: October 29, 2014,
Original Paper AMemoryEfficient Short Read De Novo Assembly Algorithm Yuki Endo 1,a) Fubito Toyama 1 Chikafumi Chiba 2 Hiroshi Mori 1 Kenji Shoji 1 Received: October 17, 2014, Accepted: October 29, 2014,
Reference & Track Manager
 Reference & Track Manager U SoftGenetics, LLC 100 Oakwood Avenue, Suite 350, State College, PA 16803 USA * info@softgenetics.com www.softgenetics.com 888-791-1270 2016 Registered Trademarks are property
Reference & Track Manager U SoftGenetics, LLC 100 Oakwood Avenue, Suite 350, State College, PA 16803 USA * info@softgenetics.com www.softgenetics.com 888-791-1270 2016 Registered Trademarks are property
Chapter 13 Reduced Instruction Set Computers
 Chapter 13 Reduced Instruction Set Computers Contents Instruction execution characteristics Use of a large register file Compiler-based register optimization Reduced instruction set architecture RISC pipelining
Chapter 13 Reduced Instruction Set Computers Contents Instruction execution characteristics Use of a large register file Compiler-based register optimization Reduced instruction set architecture RISC pipelining
Combinatorial Pattern Matching. CS 466 Saurabh Sinha
 Combinatorial Pattern Matching CS 466 Saurabh Sinha Genomic Repeats Example of repeats: ATGGTCTAGGTCCTAGTGGTC Motivation to find them: Genomic rearrangements are often associated with repeats Trace evolutionary
Combinatorial Pattern Matching CS 466 Saurabh Sinha Genomic Repeats Example of repeats: ATGGTCTAGGTCCTAGTGGTC Motivation to find them: Genomic rearrangements are often associated with repeats Trace evolutionary
Whole genome alignments using MPI-LAGAN
 Whole genome alignments using MPI-LAGAN Ruinan Zhang, Huzefa Rangwala and George Karypis Department of Computer Science & Engineering, University of Minnesota Minneapolis, MN 55455 rzhang@cs.umn.edu, rangwala@cs.umn.edu,
Whole genome alignments using MPI-LAGAN Ruinan Zhang, Huzefa Rangwala and George Karypis Department of Computer Science & Engineering, University of Minnesota Minneapolis, MN 55455 rzhang@cs.umn.edu, rangwala@cs.umn.edu,
Performance analysis of parallel de novo genome assembly in shared memory system
 IOP Conference Series: Earth and Environmental Science PAPER OPEN ACCESS Performance analysis of parallel de novo genome assembly in shared memory system To cite this article: Syam Budi Iryanto et al 2018
IOP Conference Series: Earth and Environmental Science PAPER OPEN ACCESS Performance analysis of parallel de novo genome assembly in shared memory system To cite this article: Syam Budi Iryanto et al 2018
INTRODUCTION TO CONSED
 INTRODUCTION TO CONSED OVERVIEW: Consed is a program that can be used to visually assemble and analyze sequence data. This introduction will take you through the basics of opening and operating within
INTRODUCTION TO CONSED OVERVIEW: Consed is a program that can be used to visually assemble and analyze sequence data. This introduction will take you through the basics of opening and operating within
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding. B. Majoros M. Pertea S.L. Salzberg
 Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
Efficient Implementation of a Generalized Pair HMM for Comparative Gene Finding B. Majoros M. Pertea S.L. Salzberg ab initio gene finder genome 1 MUMmer Whole-genome alignment (optional) ROSE Region-Of-Synteny
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching
 SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
3. Memory Management
 Principles of Operating Systems CS 446/646 3. Memory Management René Doursat Department of Computer Science & Engineering University of Nevada, Reno Spring 2006 Principles of Operating Systems CS 446/646
Principles of Operating Systems CS 446/646 3. Memory Management René Doursat Department of Computer Science & Engineering University of Nevada, Reno Spring 2006 Principles of Operating Systems CS 446/646
SIS: a Program to Generate Draft Genome Sequence Scaffolds for Prokaryotes Supplementary Materials
 SIS: a Program to Generate Draft Genome Sequence Scaffolds for Prokaryotes Supplementary Materials Zanoni Dias 1, Ulisses Dias 1 and João C. Setubal 2 1 Instituto de Computação, Universidade Estadual de
SIS: a Program to Generate Draft Genome Sequence Scaffolds for Prokaryotes Supplementary Materials Zanoni Dias 1, Ulisses Dias 1 and João C. Setubal 2 1 Instituto de Computação, Universidade Estadual de
KisSplice. Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data. 29th may 2013
 Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)
Identifying and Quantifying SNPs, indels and Alternative Splicing Events from RNA-seq data 29th may 2013 Next Generation Sequencing A sequencing experiment now produces millions of short reads ( 100 nt)