Advanced UCSC Browser Functions
|
|
|
- Franklin Riley
- 6 years ago
- Views:
Transcription
1 Advanced UCSC Browser Functions Dr. Thomas Randall bioinformatics.unc.edu UCSC Browser: genome.ucsc.edu
2 Overview Custom Tracks adding your own datasets Utilities custom tools for manipulating files Saving session saving alterations and custom tracks Table Browser large scale custom downloads Galaxy independent site with more custom tools, well integrated with UCSC


3 Anyone using bacterial genome Go to Archaeal browser link As example, select E coli K12

4 Custom Tracks If your own data is in the right format it can be displayed as a track
5 Custom Tracks 1) ctcfhg18.bed The file ctcfhg18.bed is adapted from the file from Cell 128: ) Nature Genetics 38: ) Cell 129: primer data 4) we will make our own bed file 1) Map primers to genome with BLAT 2) Format results in Excel to a bed file 3) Load in UCSC Browser Minimal format for a bed file Chr start stop chr chr chr chr chr chr chr chr chr chr chr chr chr
6 BED format browser extensible data BED format provides a flexible way to define the data lines that are displayed in an annotation track. BED lines have three required fields and nine additional optional fields. The number of fields per line must be consistent throughout any single set of data in an annotation track. The order of the optional fields is binding: lower-numbered fields must always be populated if higher-numbered fields are used. The first three required BED fields are: 1. chrom *- The name of the chromosome (e.g. chr3, chry, chr2_random) or scaffold (e.g. scaffold10671). 2. chromstart * - The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered chromend *- The ending position of the feature in the chromosome or scaffold. The chromend base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromstart=0, chromend=100, and span the bases numbered The 9 additional optional BED fields are: 4. name - Defines the name of the BED line. This label is displayed to the left of the BED line in the Genome Browser window when the track is open to full display mode or directly to the left of the item in pack mode. 5. score - A score between 0 and If the track line usescore attribute is set to 1 for this annotation data set, the score value will determine the level of gray in which this feature is displayed (higher numbers = darker gray). This table shows the Genome Browser's translation of BED score values into shades of gray: 6. strand - Defines the strand - either '+' or '-'. 7. thickstart - The starting position at which the feature is drawn thickly (for example, the start codon in gene displays). 8. thickend - The ending position at which the feature is drawn thickly (for example, the stop codon in gene displays). 9. itemrgb - An RGB value of the form R,G,B (e.g. 255,0,0). If the track line itemrgb attribute is set to "On", this RBG value will determine the display color of the data contained in this BED line. NOTE: It is recommended that a simple color scheme (eight colors or less) be used with this attribute to avoid overwhelming the color resources of the Genome Browser and your Internet browser. 10. blockcount - The number of blocks (exons) in the BED line. 11. blocksizes - A comma-separated list of the block sizes. The number of items in this list should correspond to blockcount. 12. blockstarts - A comma-separated list of block starts. All of the blockstart positions should be calculated relative to chromstart. The number of items in this list should correspond to blockcount.
7 File formats for Custom Tracks Bed* (already covered) BedGraph - The bedgraph format allows display of continuous-valued data in track format. This display type is useful for probability scores and transcriptome data. GFF* (General Feature Format) lines are based on the GFF standard file format. GFF lines have nine required fields that must be tab-separated. GTF (Gene Transfer Format) is a refinement to GFF that tightens the specification. WIG* - The wiggle format is for display of dense, continuous data such as GC percent, probability scores, and transcriptome data. MAF - The multiple alignment format stores a series of multiple alignments in a format that is easy to parse and relatively easy to read. This format stores multiple alignments at the DNA level between entire genomes. PSL lines represent alignments, and are typically taken from files generated by BLAT or pslayout. *most commonly seen in papers, bed for discrete data, GFF for genome annotation, WIG for chip-chip, chip-seq studies, more continuous and quantitative data

8 RGB color convention all colors are specified by some combination of values of red, green and blue from 0 to 255 In Manage Custom Tracks > Name of Track You can edit characteristics of track Add color=255,0,0 to set color as RED The Other RGB Color Chart
9 bed, gff, wig files can get BIG bed files in Cell 129: 823 over 80 Mb Example wig and gff files (big.wig and hsa.gff) on Hard to map to the UCSC Browser as sending this through the internet takes time, the link may fail Solution: local copy of the UCSC Browser, faster connection Also, if you have a big file try the Duke mirror of the UCSC Browser
10 Making your own files Always create text files as your input good general rule for all bioinformatics tools Save as plain text or tab delimited text most tools recognize empty space as a tab Create and open in Wordpad, don t use Notepad as the line breaks and delimiters are not recognized. For Macs, use Text Editor. Other text editors at
11 Building our bed file 1. Download primers.fasta 2. Open the UCSC Browser BLAT tool and input the above file. Run BLAT using default conditions. 3. Copy output of BLAT, open Excel and paste in BLAT result. 4. Select column A and under the Data header choose Text to Columns then Delimited > Next and choose space and then Next > Finish. This turns data into individual columns. 5. Eliminate all unnecessary columns (A,B,D-H,J,M). 6. Move what is now col A to col D. 7. Add chr to the chr numbers as the bed file format requires this. There are various ways to accomplish this, manually is slowest but ok for a short list. 8. Save file as tab delimited text as primers.bed to your desktop. 9. Load into custom tracks. Give a name. 10. Change color of track color=0,0,255 (blue) color=0,255, 0 (green) color=255,0,0 (red)
12 Utilities liftover - converts genome coordinates and genome annotation files between assemblies. The current version supports both forward and reverse conversions, as well as conversions between selected species. To go from hg16 to hg 18 one has to move one build at a time hg16 > hg17 > hg18 DNA and Protein duster: both removes formatting characters and other non-sequence-related characters from an input sequence. Offers several configuration options for the output format. Example: ctcf.bed (hg17) to hg18 Cell 128: 1231 (
13 Saving sessions Custom tracks persist only ~48 hrs Use Save Session to keep custom tracks and to customize Genome Browser Create an account through Sessions Sessions persist for one year from last access time To save additions to a session, over-write old session of same name Can also save session to local file and reload, send to others see HSLsession on website

14 Table Browser All data (annotation tracks) from the Genome Browser is stored, and is available through the Table Browser via custom queries
15 Tips for Table Browser First two rows important to define data table this is likely unimportant unless you are interested im MySQL databases Filter used to restrict above selection to a more defined subset Intersection used to get junctions of multiple annotation tracks Output format important to specify type of output You can generate genome wide large datasets, gzip anything you think may be large Specify a name for the file or it will be loaded into the browser window Large datasets may time out may need to go to Downloads section
16 Field descriptions for Table Browser clade: Specifies which clade the organism is in. genome: Specifies which organism data to use. assembly: Specifies which version of the organism's genome sequence to use. group: Selects the type of tracks to be displayed in the track list. The options correspond to the track groupings shown in the Genome Browser. Select 'All Tracks' for an alphabetical list of all available tracks in all groups. Select 'All Tables' to see all tables including those not associated with a track. database: (with "All Tables" group option) Determines which database should be used for options in table menu. track: Selects the annotation track data to work with. This list displays all tracks belonging to the group specified in the group list. table: Selects the SQL table data to use. This list shows all tables associated with the track specified in the track list. describe table schema: Displays schema information for the tables associated with the selected track. region: Restricts the query to a particular chromosome or region. Select genome to apply the query to the entire genome or ENCODE to examine only the ENCODE regions. To limit the query to a specific position, type a chromosome name, e.g. chrx, or a chromosome coordinate range, such as chrx: , or a gene name or other id in the text box. lookup: Press this button after typing in a gene name or other id in the position text box to look up the chromosome position identifiers (selected tracks only): Restricts the output to table data that match a list of identifiers, for instance RefSeq accessions for the RefSeq track. If no identifiers are entered, all table data within the specified region will be displayed. filter: Restricts the query to only those items that match certain criteria, e.g. genes with a single exon. Click the Create button to add a filter, the Edit button to modify an existing filter, or the Clear button to remove an existing filter. intersection (selected tracks only): Combines the output of two queries into a single set of data based on specific join criteria. For example, this can be used to find all SNPs that intersect with RefSeq coding regions. The intersection can be configured to retain the existing alignment structure of the table with a specified amount of overlap, or discard the structure in favor of a simple list of position ranges using a base-pair intersection or union of the two data sets. The button functionalities are similar to those of the filter option. output: Specifies the output format (not all options are available for some tracks). Formats include: all fields from selected table - data from the selected table displayed in a tab-separated format suitable for import into spreadsheets and relational databases. The ASCII format may be read in any web browser or text editor. selected fields from primary and related tables - user-selected set of tab-separated fields from the selected table and (optionally) other related tables as well. sequence - DNA (or protein sequence, in some cases) associated with the table. BED - positions of data items in a standard UCSC Browser format. GTF - positions of all data items in a standard gene prediction format. (Both BED and GTF formats can be used as the basis for custom tracks). CDS FASTA alignment from multiple alignment - FASTA alignments of the CDS regions of a gene prediction track using any of the multiple alignment tracks for the current database. Output sequence can be in either nucleotide-space or translated to protein-space. Available only for genepred tracks. custom track - customized Genome Browser annotation track based on the results of the query. hyperlinks to Genome Browser - returns a page full of hyperlinks to the UCSC Genome Browser, one for each item in the table. data points - the data points that make up a graph (aka wiggle) track. MAF - multiple alignments in MAF format Send output to Galaxy: displays results of query in Galaxy, a framework for interactive genome analysis. file type returned: When a filename is entered in the "output file" text box, specifies the format of the output file: plain text - data is in ASCII format gzip compressed - data is compressed in gzip format get output: Submits a data query based on the specified parameters and returns the output. summary/statistics: Displays statistics about the data specified by the parameters.
17 Downloading individual genes with the Table Browser Go to the gene in the Genome Browser Select Tables to go to Table Browser Choose position (chromosomal position of genome browser is maintained) Choose output format as sequence Give file name Choose sequence type for download (genomic, protein or mrna) Will output all sequences involved with particular track chosen if there are three RefSeqs you will get three sequences If you want a specific identifier, paste it in.
18 Example 1 basic query Dear Madam/Sir, I am intending to obtain the sequence of promoter region, about -/+ 500bp around the transcription start site. Would you please tell me how to get those sequence in batch? Thanks a lot. Best regards, Rex We will try 100 bp upstream so the download does not get too big clade: Mammal; genome: Human; assembly: Mar. 2006; group: Genes and Gene Prediction Tracks; track: UCSC Genes; table: knowngene; region: "genome", output format: "sequence" output file filename and click "get output". On the next page you can select genomic sequence and then your promotor/upstream bases. Enter 100" into each box.
19 Example 2 intersection of two datasets I want to know all the mirnas that overlap with all known mouse genes 1) Select all genes clade: Mammal; genome: Mouse; assembly:.july 2007; group: Genes and Gene Prediction Tracks; track: CCDS Genes; table: knowngene; region: "genome Create Intersection 2) Create intersection with all mirnas clade: Mammal; genome: Mouse; assembly: July 2007; group: Genes and Gene Prediction Tracks; track: mirnas; table: mirnas; use all default settings output format: "sequence set file name and click "get output". On the next page select CDS exons. RESULT 11 mouse genes overlap with mirnas
20 Example 3 filtering a dataset I want all genes on human chromosome 22 with more than 5 exons 1) Select all genes clade: Mammal; genome: Human; assembly: Mar 2006; group: Genes and Gene Prediction Tracks; track: UCSC Genes; table: knowngene; region: "genome 2) Create filter chrom does match chr22 exoncount is > 5, submit output format: "sequence set file name and click "get output". On the next page select CDS exons. RESULT 851 chr22 genes overlap with >5 exons

21 No tool is an island In genome queries, multiple tools required Interaction between tools is often the limiting factor ??? see PLOS Comp Biol 4 e121 for discussion on the interaction of these tools
22 Galaxy examples 1) Use existing ctcf18.bed file. This has coordinates for all binding sites of the CTCF protein in humans. I want the sequence corresponding to those coordinates in order to determine if there are any conserved motifs. Get Data upload file, choose species and execute Fetch sequences extract genomic DNA Save to desktop RESULT fasta formatted sequences for further motif analysis (separate tool)
23 Galaxy examples Move output of Table Browser into Galaxy for further processing. I want to know what the longest and shortest Refseq gene on chr22 are 1) In Table Browser: clade: Mammal; genome: Human; assembly: Mar 2006; group: Genes and Gene Prediction Tracks; track: Refseq Genes; table: knowngene; region: "genome Create filter 2) chrom does match chr22 Choose output to bed file and send query to Galaxy 3) In Galaxy Fetch sequences extract genomic DNA FASTA manipulation compute length Filter and Sort sort on c2, descending order


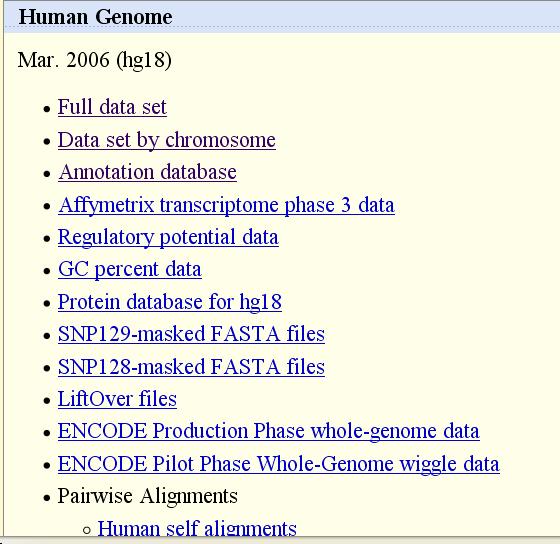
24 Downloads of all sequence data, tracks, all software
A short Introduction to UCSC Genome Browser
 A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Genome Browsers - The UCSC Genome Browser
 Genome Browsers - The UCSC Genome Browser Background The UCSC Genome Browser is a well-curated site that provides users with a view of gene or sequence information in genomic context for a specific species,
Genome Browsers - The UCSC Genome Browser Background The UCSC Genome Browser is a well-curated site that provides users with a view of gene or sequence information in genomic context for a specific species,
The UCSC Gene Sorter, Table Browser & Custom Tracks
 The UCSC Gene Sorter, Table Browser & Custom Tracks Advanced searching and discovery using the UCSC Table Browser and Custom Tracks Osvaldo Graña Bioinformatics Unit, CNIO 1 Table Browser and Custom Tracks
The UCSC Gene Sorter, Table Browser & Custom Tracks Advanced searching and discovery using the UCSC Table Browser and Custom Tracks Osvaldo Graña Bioinformatics Unit, CNIO 1 Table Browser and Custom Tracks
ChIP-Seq Tutorial on Galaxy
 1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Genome representa;on concepts. Week 12, Lecture 24. Coordinate systems. Genomic coordinates brief overview 11/13/14
 2014 - BMMB 852D: Applied Bioinforma;cs Week 12, Lecture 24 István Albert Biochemistry and Molecular Biology and Bioinforma;cs Consul;ng Center Penn State Genome representa;on concepts At the simplest
2014 - BMMB 852D: Applied Bioinforma;cs Week 12, Lecture 24 István Albert Biochemistry and Molecular Biology and Bioinforma;cs Consul;ng Center Penn State Genome representa;on concepts At the simplest
Genomics 92 (2008) Contents lists available at ScienceDirect. Genomics. journal homepage:
 Contents lists available at ScienceDirect. Genomics. journal homepage:") Genomics 92 (2008) 75 84 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Review UCSC genome browser tutorial Ann S. Zweig a,, Donna Karolchik a, Robert
Genomics 92 (2008) 75 84 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Review UCSC genome browser tutorial Ann S. Zweig a,, Donna Karolchik a, Robert
Tutorial 1: Exploring the UCSC Genome Browser
 Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Genomic Analysis with Genome Browsers.
 Genomic Analysis with Genome Browsers http://barc.wi.mit.edu/hot_topics/ 1 Outline Genome browsers overview UCSC Genome Browser Navigating: View your list of regions in the browser Available tracks (eg.
Genomic Analysis with Genome Browsers http://barc.wi.mit.edu/hot_topics/ 1 Outline Genome browsers overview UCSC Genome Browser Navigating: View your list of regions in the browser Available tracks (eg.
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
Genome Browsers Guide
 Genome Browsers Guide Take a Class This guide supports the Galter Library class called Genome Browsers. See our Classes schedule for the next available offering. If this class is not on our upcoming schedule,
Genome Browsers Guide Take a Class This guide supports the Galter Library class called Genome Browsers. See our Classes schedule for the next available offering. If this class is not on our upcoming schedule,
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
Useful software utilities for computational genomics. Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017
 Useful software utilities for computational genomics Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017 Overview Search and download genomic datasets: GEOquery, GEOsearch and GEOmetadb,
Useful software utilities for computational genomics Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017 Overview Search and download genomic datasets: GEOquery, GEOsearch and GEOmetadb,
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
The UCSC Genome Browser
 The UCSC Genome Browser Search, retrieve and display the data that you want Materials prepared by Warren C. Lathe, Ph.D. Mary Mangan, Ph.D. www.openhelix.com Updated: Q3 2006 Version_0906 Copyright OpenHelix.
The UCSC Genome Browser Search, retrieve and display the data that you want Materials prepared by Warren C. Lathe, Ph.D. Mary Mangan, Ph.D. www.openhelix.com Updated: Q3 2006 Version_0906 Copyright OpenHelix.
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
- 1 - Web page:
 J-Circos Manual 2014-11-10 J-Circos: A Java Graphic User Interface for Circos Plot Jiyuan An 1, John Lai 1, Atul Sajjanhar 2, Jyotsna Batra 1,Chenwei Wang 1 and Colleen C Nelson 1 1 Australian Prostate
J-Circos Manual 2014-11-10 J-Circos: A Java Graphic User Interface for Circos Plot Jiyuan An 1, John Lai 1, Atul Sajjanhar 2, Jyotsna Batra 1,Chenwei Wang 1 and Colleen C Nelson 1 1 Australian Prostate
User's guide to ChIP-Seq applications: command-line usage and option summary
 User's guide to ChIP-Seq applications: command-line usage and option summary 1. Basics about the ChIP-Seq Tools The ChIP-Seq software provides a set of tools performing common genome-wide ChIPseq analysis
User's guide to ChIP-Seq applications: command-line usage and option summary 1. Basics about the ChIP-Seq Tools The ChIP-Seq software provides a set of tools performing common genome-wide ChIPseq analysis
Introduction to Genome Browsers
 Introduction to Genome Browsers Rolando Garcia-Milian, MLS, AHIP (Rolando.milian@ufl.edu) Department of Biomedical and Health Information Services Health Sciences Center Libraries, University of Florida
Introduction to Genome Browsers Rolando Garcia-Milian, MLS, AHIP (Rolando.milian@ufl.edu) Department of Biomedical and Health Information Services Health Sciences Center Libraries, University of Florida
The UCSC Genome Browser
 The UCSC Genome Browser UNIT 1.4 The rapid progress of public sequencing and mapping efforts on vertebrate genomes has increased the demand for tools that offer quick and easy access to the data at many
The UCSC Genome Browser UNIT 1.4 The rapid progress of public sequencing and mapping efforts on vertebrate genomes has increased the demand for tools that offer quick and easy access to the data at many
UCSC Genome Browser ASHG 2014 Workshop
 UCSC Genome Browser ASHG 2014 Workshop We will be using human assembly hg19. Some steps may seem a bit cryptic or truncated. That is by design, so you will think about things as you go. In this document,
UCSC Genome Browser ASHG 2014 Workshop We will be using human assembly hg19. Some steps may seem a bit cryptic or truncated. That is by design, so you will think about things as you go. In this document,
Genetics 211 Genomics Winter 2014 Problem Set 4
 Genomics - Part 1 due Friday, 2/21/2014 by 9:00am Part 2 due Friday, 3/7/2014 by 9:00am For this problem set, we re going to use real data from a high-throughput sequencing project to look for differential
Genomics - Part 1 due Friday, 2/21/2014 by 9:00am Part 2 due Friday, 3/7/2014 by 9:00am For this problem set, we re going to use real data from a high-throughput sequencing project to look for differential
Analyzing Variant Call results using EuPathDB Galaxy, Part II
 Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
BovineMine Documentation
 BovineMine Documentation Release 1.0 Deepak Unni, Aditi Tayal, Colin Diesh, Christine Elsik, Darren Hag Oct 06, 2017 Contents 1 Tutorial 3 1.1 Overview.................................................
BovineMine Documentation Release 1.0 Deepak Unni, Aditi Tayal, Colin Diesh, Christine Elsik, Darren Hag Oct 06, 2017 Contents 1 Tutorial 3 1.1 Overview.................................................
Introduction to Galaxy
 Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
From genomic regions to biology
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
UCSC Genome Browser Pittsburgh Workshop -- Practical Exercises
 UCSC Genome Browser Pittsburgh Workshop -- Practical Exercises We will be using human assembly hg19. These problems will take you through a variety of resources at the UCSC Genome Browser. You will learn
UCSC Genome Browser Pittsburgh Workshop -- Practical Exercises We will be using human assembly hg19. These problems will take you through a variety of resources at the UCSC Genome Browser. You will learn
Using the UCSC genome browser
 Using the UCSC genome browser Credits Terry Braun Mary Mangan, Ph.D. www.openhelix.com UCSC Genome Browser Credits Development team: http://genome.ucsc.edu/staff.html n Led by David Haussler and Jim Kent
Using the UCSC genome browser Credits Terry Braun Mary Mangan, Ph.D. www.openhelix.com UCSC Genome Browser Credits Development team: http://genome.ucsc.edu/staff.html n Led by David Haussler and Jim Kent
Practical Course in Genome Bioinformatics
 Practical Course in Genome Bioinformatics 20/01/2017 Exercises - Day 1 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2017/ Answer questions Q1-Q3 below and include requested Figures 1-5
Practical Course in Genome Bioinformatics 20/01/2017 Exercises - Day 1 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2017/ Answer questions Q1-Q3 below and include requested Figures 1-5
4.1. Access the internet and log on to the UCSC Genome Bioinformatics Web Page (Figure 1-
 1. PURPOSE To provide instructions for finding rs Numbers (SNP database ID numbers) and increasing sequence length by utilizing the UCSC Genome Bioinformatics Database. 2. MATERIALS 2.1. Sequence Information
1. PURPOSE To provide instructions for finding rs Numbers (SNP database ID numbers) and increasing sequence length by utilizing the UCSC Genome Bioinformatics Database. 2. MATERIALS 2.1. Sequence Information
The UCSC Genome Browser: What Every Molecular Biologist Should Know
 The UCSC Genome Browser: What Every Molecular Biologist Should Know Mary E. Mangan, 1 Jennifer M. Williams, 1 Robert M. Kuhn, 2 and Warren C. Lathe III 1 UNIT 19.9 1 OpenHelix LLC, Bellevue, Washington
The UCSC Genome Browser: What Every Molecular Biologist Should Know Mary E. Mangan, 1 Jennifer M. Williams, 1 Robert M. Kuhn, 2 and Warren C. Lathe III 1 UNIT 19.9 1 OpenHelix LLC, Bellevue, Washington
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Goldmine: Integrating information to place sets of genomic ranges into biological contexts Jeff Bhasin
 Goldmine: Integrating information to place sets of genomic ranges into biological contexts Jeff Bhasin 2016-04-22 Contents Purpose 1 Prerequisites 1 Installation 2 Example 1: Annotation of Any Set of Genomic
Goldmine: Integrating information to place sets of genomic ranges into biological contexts Jeff Bhasin 2016-04-22 Contents Purpose 1 Prerequisites 1 Installation 2 Example 1: Annotation of Any Set of Genomic
Click on "+" button Select your VCF data files (see #Input Formats->1 above) Remove file from files list:
 Remove file from files list:") CircosVCF: CircosVCF is a web based visualization tool of genome-wide variant data described in VCF files using circos plots. The provided visualization capabilities, gives a broad overview of the genomic
CircosVCF: CircosVCF is a web based visualization tool of genome-wide variant data described in VCF files using circos plots. The provided visualization capabilities, gives a broad overview of the genomic
Sequence Alignment. GBIO0002 Archana Bhardwaj University of Liege
 Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
The BEDTools manual. Last updated: 21-September-2010 Current as of BEDTools version Aaron R. Quinlan and Ira M. Hall University of Virginia
 The BEDTools manual Last updated: 21-September-2010 Current as of BEDTools version 2.10.0 Aaron R. Quinlan and Ira M. Hall University of Virginia Contact: aaronquinlan@gmail.com 1. OVERVIEW... 7 1.1 BACKGROUND...7
The BEDTools manual Last updated: 21-September-2010 Current as of BEDTools version 2.10.0 Aaron R. Quinlan and Ira M. Hall University of Virginia Contact: aaronquinlan@gmail.com 1. OVERVIEW... 7 1.1 BACKGROUND...7
Browser Exercises - I. Alignments and Comparative genomics
 Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Browser Exercises - I Alignments and Comparative genomics 1. Navigating to the Genome Browser (GBrowse) Note: For this exercise use http://www.tritrypdb.org a. Navigate to the Genome Browser (GBrowse)
Design and Annotation Files
 Design and Annotation Files Release Notes SeqCap EZ Exome Target Enrichment System The design and annotation files provide information about genomic regions covered by the capture probes and the genes
Design and Annotation Files Release Notes SeqCap EZ Exome Target Enrichment System The design and annotation files provide information about genomic regions covered by the capture probes and the genes
Data Walkthrough: Background
 Data Walkthrough: Background File Types FASTA Files FASTA files are text-based representations of genetic information. They can contain nucleotide or amino acid sequences. For this activity, students will
Data Walkthrough: Background File Types FASTA Files FASTA files are text-based representations of genetic information. They can contain nucleotide or amino acid sequences. For this activity, students will
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
Exercise 2: Browser-Based Annotation and RNA-Seq Data
 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
The UCSC Genome Browser
 The UCSC Genome Browser Donna Karolchik, 1 Angie S. Hinrichs, 1 and W. James Kent 1 UNIT 1.4 1 Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, California
The UCSC Genome Browser Donna Karolchik, 1 Angie S. Hinrichs, 1 and W. James Kent 1 UNIT 1.4 1 Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, California
Public Repositories Tutorial: Bulk Downloads
 Public Repositories Tutorial: Bulk Downloads Almost all of the public databases, genome browsers, and other tools you have explored so far offer some form of access to rapidly download all or large chunks
Public Repositories Tutorial: Bulk Downloads Almost all of the public databases, genome browsers, and other tools you have explored so far offer some form of access to rapidly download all or large chunks
Creating and Using Genome Assemblies Tutorial
 Creating and Using Genome Assemblies Tutorial Release 8.1 Golden Helix, Inc. March 18, 2014 Contents 1. Create a Genome Assembly for Danio rerio 2 2. Building Annotation Sources 5 A. Creating a Reference
Creating and Using Genome Assemblies Tutorial Release 8.1 Golden Helix, Inc. March 18, 2014 Contents 1. Create a Genome Assembly for Danio rerio 2 2. Building Annotation Sources 5 A. Creating a Reference
Tutorial: chloroplast genomes
 Tutorial: chloroplast genomes Stacia Wyman Department of Computer Sciences Williams College Williamstown, MA 01267 March 10, 2005 ASSUMPTIONS: You are using Internet Explorer under OS X on the Mac. You
Tutorial: chloroplast genomes Stacia Wyman Department of Computer Sciences Williams College Williamstown, MA 01267 March 10, 2005 ASSUMPTIONS: You are using Internet Explorer under OS X on the Mac. You
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
How to use earray to create custom content for the SureSelect Target Enrichment platform. Page 1
 How to use earray to create custom content for the SureSelect Target Enrichment platform Page 1 Getting Started Access earray Access earray at: https://earray.chem.agilent.com/earray/ Log in to earray,
How to use earray to create custom content for the SureSelect Target Enrichment platform Page 1 Getting Started Access earray Access earray at: https://earray.chem.agilent.com/earray/ Log in to earray,
HymenopteraMine Documentation
 HymenopteraMine Documentation Release 1.0 Aditi Tayal, Deepak Unni, Colin Diesh, Chris Elsik, Darren Hagen Apr 06, 2017 Contents 1 Welcome to HymenopteraMine 3 1.1 Overview of HymenopteraMine.....................................
HymenopteraMine Documentation Release 1.0 Aditi Tayal, Deepak Unni, Colin Diesh, Chris Elsik, Darren Hagen Apr 06, 2017 Contents 1 Welcome to HymenopteraMine 3 1.1 Overview of HymenopteraMine.....................................
ChIP-seq practical: peak detection and peak annotation. Mali Salmon-Divon Remco Loos Myrto Kostadima
 ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
Today's outline. Resources. Genome browser components. Genome browsers: Discovering biology through genomics. Genome browser tutorial materials
 Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Welcome to GenomeView 101!
 Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
The BEDTools manual. Aaron R. Quinlan and Ira M. Hall University of Virginia. Contact:
 The BEDTools manual Aaron R. Quinlan and Ira M. Hall University of Virginia Contact: aaronquinlan@gmail.com 1. OVERVIEW... 6 1.1 BACKGROUND...6 1.2 SUMMARY OF AVAILABLE TOOLS...7 1.3 FUNDAMENTAL CONCEPTS
The BEDTools manual Aaron R. Quinlan and Ira M. Hall University of Virginia Contact: aaronquinlan@gmail.com 1. OVERVIEW... 6 1.1 BACKGROUND...6 1.2 SUMMARY OF AVAILABLE TOOLS...7 1.3 FUNDAMENTAL CONCEPTS
A manual for the use of mirvas
 A manual for the use of mirvas Authors: Sophia Cammaerts, Mojca Strazisar, Jenne Dierckx, Jurgen Del Favero, Peter De Rijk Version: 1.0.2 Date: July 27, 2015 Contact: peter.derijk@gmail.com, mirvas.software@gmail.com
A manual for the use of mirvas Authors: Sophia Cammaerts, Mojca Strazisar, Jenne Dierckx, Jurgen Del Favero, Peter De Rijk Version: 1.0.2 Date: July 27, 2015 Contact: peter.derijk@gmail.com, mirvas.software@gmail.com
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
ChromHMM: automating chromatin-state discovery and characterization
 Nature Methods ChromHMM: automating chromatin-state discovery and characterization Jason Ernst & Manolis Kellis Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure 3 Supplementary Figure
Nature Methods ChromHMM: automating chromatin-state discovery and characterization Jason Ernst & Manolis Kellis Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure 3 Supplementary Figure
CisGenome User s Manual
 CisGenome User s Manual 1. Overview 1.1 Basic Framework of CisGenome 1.2 Installation 1.3 Summary of All Functions 1.4 A Quick Start Analysis of a ChIP-chip Experiment 2. Genomics Toolbox I Establishing
CisGenome User s Manual 1. Overview 1.1 Basic Framework of CisGenome 1.2 Installation 1.3 Summary of All Functions 1.4 A Quick Start Analysis of a ChIP-chip Experiment 2. Genomics Toolbox I Establishing
panda Documentation Release 1.0 Daniel Vera
 panda Documentation Release 1.0 Daniel Vera February 12, 2014 Contents 1 mat.make 3 1.1 Usage and option summary....................................... 3 1.2 Arguments................................................
panda Documentation Release 1.0 Daniel Vera February 12, 2014 Contents 1 mat.make 3 1.1 Usage and option summary....................................... 3 1.2 Arguments................................................
Annotating a single sequence
 BioNumerics Tutorial: Annotating a single sequence 1 Aim The annotation application in BioNumerics has been designed for the annotation of coding regions on sequences. In this tutorial you will learn how
BioNumerics Tutorial: Annotating a single sequence 1 Aim The annotation application in BioNumerics has been designed for the annotation of coding regions on sequences. In this tutorial you will learn how
When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame
 1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
Supplementary Figure 1. Fast read-mapping algorithm of BrowserGenome.
 Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
epigenomegateway.wustl.edu
 Everything can be found at epigenomegateway.wustl.edu REFERENCES 1. Zhou X, et al., Nature Methods 8, 989-990 (2011) 2. Zhou X & Wang T, Current Protocols in Bioinformatics Unit 10.10 (2012) 3. Zhou X,
Everything can be found at epigenomegateway.wustl.edu REFERENCES 1. Zhou X, et al., Nature Methods 8, 989-990 (2011) 2. Zhou X & Wang T, Current Protocols in Bioinformatics Unit 10.10 (2012) 3. Zhou X,
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
HIPPIE User Manual. (v0.0.2-beta, 2015/4/26, Yih-Chii Hwang, yihhwang [at] mail.med.upenn.edu)
![HIPPIE User Manual. (v0.0.2-beta, 2015/4/26, Yih-Chii Hwang, yihhwang [at] mail.med.upenn.edu)](/thumbs/71/65752105.jpg "HIPPIE User Manual. (v0.0.2-beta, 2015/4/26, Yih-Chii Hwang, yihhwang [at] mail.med.upenn.edu)") HIPPIE User Manual (v0.0.2-beta, 2015/4/26, Yih-Chii Hwang, yihhwang [at] mail.med.upenn.edu) OVERVIEW OF HIPPIE o Flowchart of HIPPIE o Requirements PREPARE DIRECTORY STRUCTURE FOR HIPPIE EXECUTION o
HIPPIE User Manual (v0.0.2-beta, 2015/4/26, Yih-Chii Hwang, yihhwang [at] mail.med.upenn.edu) OVERVIEW OF HIPPIE o Flowchart of HIPPIE o Requirements PREPARE DIRECTORY STRUCTURE FOR HIPPIE EXECUTION o
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
Using Galaxy to Perform Large-Scale Interactive Data Analyses
 Using Galaxy to Perform Large-Scale Interactive Data Analyses Jennifer Hillman-Jackson, 1 Dave Clements, 2 Daniel Blankenberg, 1 James Taylor, 2 Anton Nekrutenko, 1 and Galaxy Team 1,2 UNIT 10.5 1 Penn
Using Galaxy to Perform Large-Scale Interactive Data Analyses Jennifer Hillman-Jackson, 1 Dave Clements, 2 Daniel Blankenberg, 1 James Taylor, 2 Anton Nekrutenko, 1 and Galaxy Team 1,2 UNIT 10.5 1 Penn
Tutorial 4 BLAST Searching the CHO Genome
 Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Briefly: Bioinformatics File Formats. J Fass September 2018
 Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
KGBassembler Manual. A Karyotype-based Genome Assembler for Brassicaceae Species. Version 1.2. August 16 th, 2012
 KGBassembler Manual A Karyotype-based Genome Assembler for Brassicaceae Species Version 1.2 August 16 th, 2012 Authors: Chuang Ma, Hao Chen, Mingming Xin, Ruolin Yang and Xiangfeng Wang Contact: Dr. Xiangfeng
KGBassembler Manual A Karyotype-based Genome Assembler for Brassicaceae Species Version 1.2 August 16 th, 2012 Authors: Chuang Ma, Hao Chen, Mingming Xin, Ruolin Yang and Xiangfeng Wang Contact: Dr. Xiangfeng
User Manual. Ver. 3.0 March 19, 2012
 User Manual Ver. 3.0 March 19, 2012 Table of Contents 1. Introduction... 2 1.1 Rationale... 2 1.2 Software Work-Flow... 3 1.3 New in GenomeGems 3.0... 4 2. Software Description... 5 2.1 Key Features...
User Manual Ver. 3.0 March 19, 2012 Table of Contents 1. Introduction... 2 1.1 Rationale... 2 1.2 Software Work-Flow... 3 1.3 New in GenomeGems 3.0... 4 2. Software Description... 5 2.1 Key Features...
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
Copy Number Variations Detection - TD. Using Sequenza under Galaxy
 Copy Number Variations Detection - TD Using Sequenza under Galaxy I. Data loading We will analyze the copy number variations of a human tumor (parotid gland carcinoma), limited to the chr17, from a WES
Copy Number Variations Detection - TD Using Sequenza under Galaxy I. Data loading We will analyze the copy number variations of a human tumor (parotid gland carcinoma), limited to the chr17, from a WES
Finding and Exporting Data. BioMart
 September 2017 Finding and Exporting Data Not sure what tool to use to find and export data? BioMart is used to retrieve data for complex queries, involving a few or many genes or even complete genomes.
September 2017 Finding and Exporting Data Not sure what tool to use to find and export data? BioMart is used to retrieve data for complex queries, involving a few or many genes or even complete genomes.
Easy visualization of the read coverage using the CoverageView package
 Easy visualization of the read coverage using the CoverageView package Ernesto Lowy European Bioinformatics Institute EMBL June 13, 2018 > options(width=40) > library(coverageview) 1 Introduction This
Easy visualization of the read coverage using the CoverageView package Ernesto Lowy European Bioinformatics Institute EMBL June 13, 2018 > options(width=40) > library(coverageview) 1 Introduction This
Part 1: How to use IGV to visualize variants
 Using IGV to identify true somatic variants from the false variants http://www.broadinstitute.org/igv A FAQ, sample files and a user guide are available on IGV website If you use IGV in your publication:
Using IGV to identify true somatic variants from the false variants http://www.broadinstitute.org/igv A FAQ, sample files and a user guide are available on IGV website If you use IGV in your publication:
Ion AmpliSeq Designer: Getting Started
 Ion AmpliSeq Designer: Getting Started USER GUIDE Publication Number MAN0010907 Revision F.0 For Research Use Only. Not for use in diagnostic procedures. Manufacturer: Life Technologies Corporation Carlsbad,
Ion AmpliSeq Designer: Getting Started USER GUIDE Publication Number MAN0010907 Revision F.0 For Research Use Only. Not for use in diagnostic procedures. Manufacturer: Life Technologies Corporation Carlsbad,
Tour Guide for Windows and Macintosh
 Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
Tour Guide for Windows and Macintosh 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Suite 100A, Ann Arbor, MI 48108 USA phone 1.800.497.4939 or 1.734.769.7249 (fax) 1.734.769.7074
You will be re-directed to the following result page.
 ENCODE Element Browser Goal: to navigate the candidate DNA elements predicted by the ENCODE consortium, including gene expression, DNase I hypersensitive sites, TF binding sites, and candidate enhancers/promoters.
ENCODE Element Browser Goal: to navigate the candidate DNA elements predicted by the ENCODE consortium, including gene expression, DNase I hypersensitive sites, TF binding sites, and candidate enhancers/promoters.
Annotating sequences in batch
 BioNumerics Tutorial: Annotating sequences in batch 1 Aim The annotation application in BioNumerics has been designed for the annotation of coding regions on sequences. In this tutorial you will learn
BioNumerics Tutorial: Annotating sequences in batch 1 Aim The annotation application in BioNumerics has been designed for the annotation of coding regions on sequences. In this tutorial you will learn
For Research Use Only. Not for use in diagnostic procedures.
 SMRT View Guide For Research Use Only. Not for use in diagnostic procedures. P/N 100-088-600-02 Copyright 2012, Pacific Biosciences of California, Inc. All rights reserved. Information in this document
SMRT View Guide For Research Use Only. Not for use in diagnostic procedures. P/N 100-088-600-02 Copyright 2012, Pacific Biosciences of California, Inc. All rights reserved. Information in this document
Table of contents Genomatix AG 1
 Table of contents! Introduction! 3 Getting started! 5 The Genome Browser window! 9 The toolbar! 9 The general annotation tracks! 12 Annotation tracks! 13 The 'Sequence' track! 14 The 'Position' track!
Table of contents! Introduction! 3 Getting started! 5 The Genome Browser window! 9 The toolbar! 9 The general annotation tracks! 12 Annotation tracks! 13 The 'Sequence' track! 14 The 'Position' track!
Simulation of Molecular Evolution with Bioinformatics Analysis
 Simulation of Molecular Evolution with Bioinformatics Analysis Barbara N. Beck, Rochester Community and Technical College, Rochester, MN Project created by: Barbara N. Beck, Ph.D., Rochester Community
Simulation of Molecular Evolution with Bioinformatics Analysis Barbara N. Beck, Rochester Community and Technical College, Rochester, MN Project created by: Barbara N. Beck, Ph.D., Rochester Community
Topics of the talk. Biodatabases. Data types. Some sequence terminology...
 Topics of the talk Biodatabases Jarno Tuimala / Eija Korpelainen CSC What data are stored in biological databases? What constitutes a good database? Nucleic acid sequence databases Amino acid sequence
Topics of the talk Biodatabases Jarno Tuimala / Eija Korpelainen CSC What data are stored in biological databases? What constitutes a good database? Nucleic acid sequence databases Amino acid sequence
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature25174 Sequences of DNA primers used in this study. Primer Primer sequence code 498 GTCCAGATCTTGATTAAGAAAAATGAAGAAA F pegfp 499 GTCCAGATCTTGGTTAAGAAAAATGAAGAAA F pegfp 500 GTCCCTGCAGCCTAGAGGGTTAGG
doi:10.1038/nature25174 Sequences of DNA primers used in this study. Primer Primer sequence code 498 GTCCAGATCTTGATTAAGAAAAATGAAGAAA F pegfp 499 GTCCAGATCTTGGTTAAGAAAAATGAAGAAA F pegfp 500 GTCCCTGCAGCCTAGAGGGTTAGG
Fusion Detection Using QIAseq RNAscan Panels
 Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Agilent Genomic Workbench Lite Edition 6.5
 Agilent Genomic Workbench Lite Edition 6.5 SureSelect Quality Analyzer User Guide For Research Use Only. Not for use in diagnostic procedures. Agilent Technologies Notices Agilent Technologies, Inc. 2010
Agilent Genomic Workbench Lite Edition 6.5 SureSelect Quality Analyzer User Guide For Research Use Only. Not for use in diagnostic procedures. Agilent Technologies Notices Agilent Technologies, Inc. 2010
MetScape User Manual
 MetScape 2.3.2 User Manual A Plugin for Cytoscape National Center for Integrative Biomedical Informatics July 2012 2011 University of Michigan This work is supported by the National Center for Integrative
MetScape 2.3.2 User Manual A Plugin for Cytoscape National Center for Integrative Biomedical Informatics July 2012 2011 University of Michigan This work is supported by the National Center for Integrative
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6
 Exercise 6") Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
Analysis of ChIP-seq data
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
GROK User Guide August 28, 2013
 GROK User Guide August 28, 2013 Kristian Ovaska Contact: kristian.ovaska@helsinki.fi Biomedicum Helsinki, University of Helsinki, Finland Contents 1 Introduction 1 2 Core concepts: region and region store
GROK User Guide August 28, 2013 Kristian Ovaska Contact: kristian.ovaska@helsinki.fi Biomedicum Helsinki, University of Helsinki, Finland Contents 1 Introduction 1 2 Core concepts: region and region store
Genome Browser. Background & Strategy. Spring 2017 Faction II
 Genome Browser Background & Strategy Spring 2017 Faction II Outline Beginning of the Last Phase Goals State of Art Applicable Genome Browsers Not So Genome Browsers Storing Data Strategy for the website
Genome Browser Background & Strategy Spring 2017 Faction II Outline Beginning of the Last Phase Goals State of Art Applicable Genome Browsers Not So Genome Browsers Storing Data Strategy for the website
Handling genomic data using Bioconductor II: GenomicRanges and GenomicFeatures
 Handling genomic data using Bioconductor II: GenomicRanges and GenomicFeatures Motivating examples Genomic Features (e.g., genes, exons, CpG islands) on the genome are often represented as intervals, e.g.,
Handling genomic data using Bioconductor II: GenomicRanges and GenomicFeatures Motivating examples Genomic Features (e.g., genes, exons, CpG islands) on the genome are often represented as intervals, e.g.,
Background and Strategy. Smitha, Adrian, Devin, Jeff, Ali, Sanjeev, Karthikeyan
 Background and Strategy Smitha, Adrian, Devin, Jeff, Ali, Sanjeev, Karthikeyan What is a genome browser? A web/desktop based graphical tool for rapid and reliable display of any requested portion of the
Background and Strategy Smitha, Adrian, Devin, Jeff, Ali, Sanjeev, Karthikeyan What is a genome browser? A web/desktop based graphical tool for rapid and reliable display of any requested portion of the