Analysis of ChIP-seq data
|
|
|
- Job Perry
- 5 years ago
- Views:
Transcription
1 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and open the folder you created 1
2 Analysis of ChIP-seq data BaRC Hot Topics March
3 Outline ChIP-seq overview Quality control Experimental design Mapping Map reads Remove unmapped reads and convert to bam files Check the profile of the mapped reads Peak calling 3

4 ChIP-Seq overview Park, P. J., ChIP-seq: advantages and challenges of a maturing technology, Nat Rev Genet. Oct;10(10): (2009) 4
5 Steps in data analysis 1. Quality control 2. Effective mapping Treat IP and control the same way (preprocessing and mapping) 3. Peak calling i) Read extension and signal profile generation ii) Peak assignment Pepke, S. et al. Computation for ChIP-seq and RNA-seq studies, Nat Methods. Nov (2009) 5
6 Experimental design Include a control sample. If the protein of interest binds repetitive regions using paired end sequencing may reduce the mapping ambiguity. Otherwise single reads should be fine. Include at least two biological replicates. If you have replicas you may want to use the parameter IDR irreproducible discovery rate. Talk to us if you want to learn more about it. If only a small percentage of the reads maps to the genome, you may have to troubleshot your ChIP protocol. 6
7 Data that we will use on the hands on Data is from ENCODE the exercises EncodeBroadHistoneHepg2H3k4me3 EncodeBroadHistoneHepg2Control For Quality control and mapping we will use a subset of the data (10%) For running MACS we will use the full set but taking only reads mapped to chr1 7
8 Quality control (before read mapping) Quality Control and Mapping Reads Run fastqc Look at output and determine 1. What type of quality encoding (Illumina versions) your reads have Input qualities --solexa-quals <= phred phred33 >= 1.8 Illumina versions 2. If you need to remove adapter, trim reads, filter reads based on quality etc. 8
9 Hands on Step 1: Quality control Run step 1 commands (fastqc) bsub fastqc Hepg2Control_subset.fastq bsub fastqc Hepg2H3k4me3_subset.fastq 9

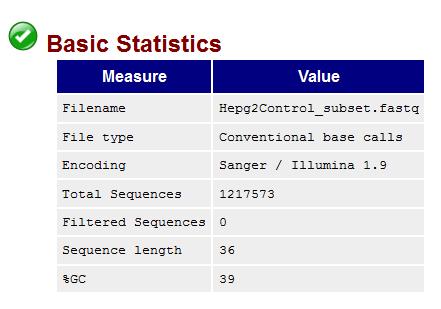
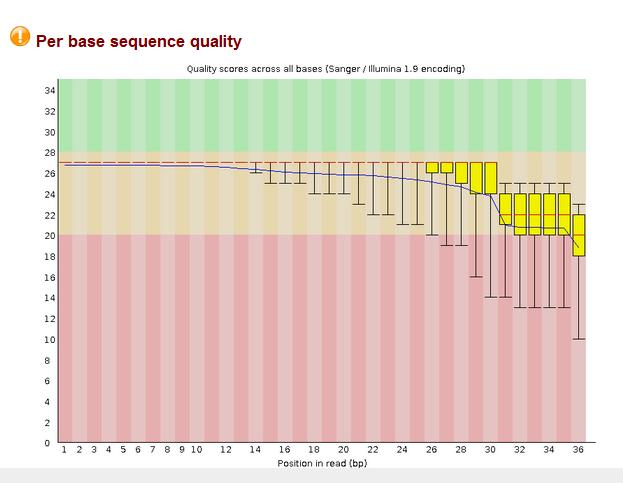
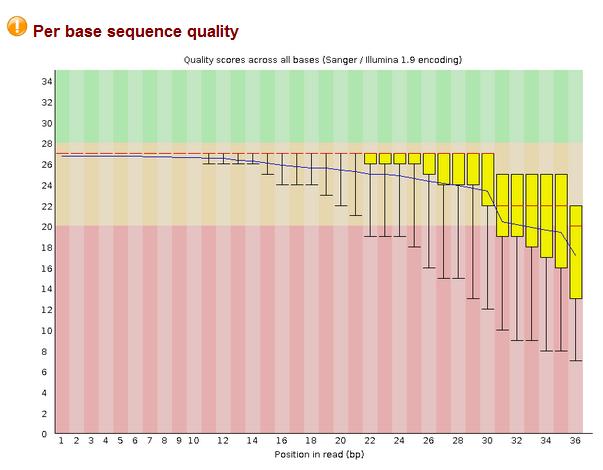
10 Fastqc Output 10
11 Mapping We have to map reads to the genome. The mapping software doesn t have to know about splicing. Bowtie, bowtie2 (allows gaps) are good choices Bwa (allows gaps) is fine too Treat IP and control samples the same way during preprocessing and mapping. 11
12 Hands on Step 2: Mapping bsub bowtie -k 1 -n 2 -l 36 --best --sam --phred33-quals /nfs/genomes/human_gp_feb_09_no_random/bowtie/hg19 Hepg2Control_subset.fastq Hepg2Control_subset_hg19.k1.n2.l36.best.sam bsub bowtie -k 1 -n 2 -l 36 --best --sam --phred33-quals /nfs/genomes/human_gp_feb_09_no_random/bowtie/hg19 Hepg2H3k4me3_subset.fastq Hepg2H3k4me3_subset_hg19.k1.n2.l36.best.sam --phred33-quals Because we have Illumina 1.9 -k 1 report up to 1 good alignments per read (this is the default setting) -n 2 max mismatches in seed (this is the default setting) -l 36 seed length (make this the same or close to the read length) --best report the best alignment 12
13 Hands on Steps 3: Remove unmapped reads and convert to BAM This step makes the final files smaller. bsub "samtools view -hs -F 4 Hepg2Control_subset_hg19.k1.n2.l36.best.sam samtools view -b -S - > Hepg2Control_subset_hg19.k1.n2.l36.best.mapped_only.bam bsub "samtools view -hs -F 4 Hepg2H3k4me3_subset_hg19.k1.n2.l36.best.sam samtools view -b -S - > Hepg2H3k4me3_subset_hg19.k1.n2.l36.best.mapped_only.bam" 13
14 Hands on Steps 4: Quality control after read mapping Check the strand cross-correlation peak and predominant fragment length. bsub Rscript /nfs/barc_public/phantompeakqualtools/run_spp.r -c=control_chr1.bam -savp -out=control_chr1.run_spp.out bsub Rscript /nfs/barc_public/phantompeakqualtools/run_spp.r -c=hepg2h3k4me3_chr1.bam -savp -out=hepg2h3k4me3_chr1.run_spp.out -savp, save cross-correlation plot 14
15 Strand cross-correlation analysis Bailey, et al. Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data, PLOS Computational Biology Nov 2013 Strand cross-correlation is computed as the Pearson correlation between the positive and the negative strand profiles at different strand shift distances, k 15
16 Strand cross-correlation analysis outputs 16
17 Peak calling Peak calling i) Read extension and signal profile generation strand cross-correlation can be used to calculate fragment length ii) Peak assignment Look for fold enrichment of the sample over input or expected background Estimate the significance of the fold enrichment using: Poisson distribution negative binomial distribution background distribution from input DNA model background data to adjust for local variation (MACS) Pepke, S. et al. Computation for ChIP-seq and RNA-seq studies, Nat Methods. Nov
18 Peak calling: MACS MACS uses a two-step strategy: 1. Builds a model using high quality peaks and infers shift size from it. 2. Shifts the reads and does a second round to call the final peaks. It uses a Poisson distribution to assign p-values to peaks. But the distribution has a dynamic parameter, local lambda, to capture the influence of local biases. MACS default is to filter out redundant tags at the same location and with the same strand by allowing at most 1 tag. This works well. -g: You need to set up this parameter accordingly: Effective genome size. It can be 1.0e+9 or , or shortcuts: 'hs' for human (2.7e9), 'mm' for mouse (1.87e9), 'ce' for C. elegans (9e7) and 'dm' for fruit fly (1.2e8), Default:hs For broad peaks like some histone modifications it is recommended to use --nomodel and if there is not input sample to use --nolambda. 18
19 Hands on Steps 5: MACS Input files: Hepg2H3k4me3_chr1.bam Control_chr1.bam MACS command bsub "macs -t Hepg2H3k4me3_chr1.bam -c Control_chr1.bam --name=hepg2h3k4me3_chr1 --format=bam --wig --space=25 " PARAMETERS -t TFILE Treatment file -c CFILE Control file --name=name Experiment name, which will be used to generate output file names. DEFAULT: NA --format=format Format of tag file, BED or ELAND or ELANDMULTI or ELANDMULTIPET or SAM or BAM or BOWTIE. DEFAULT: BED --wig: Whether or not to save shifted raw tag count at every bp into a wiggle file --space=space The resolution for saving wiggle files, by default, MACS will save the raw tag count every 10 bps. Usable only with '--wig' option 19
20 MACS output Output files: 1. Folder with wig files for control and sample. 2. Excel file for peaks found containing the following columns : chr->start ->end->length->summit->tags-> -10*LOG10(pvalue) ->fold_enrichment->fdr(%) 3. Bed file with peaks found: chr->start->end-> -10*LOG10(pvalue) 4. Excel file for negative peaks Looking at the output on a genome browser: To visualize the peaks you can upload the bed file into a genome browser. You can also make a bedgraph file with columns (step 6 of hands on): chr->start ->end->fold_enrichment 20
21 Visualize your peaks in IGV Treatment wig Control wig Peaks bedgraph Peaks bed RefSeq Genes 21
22 Other recommendations Look at your raw data as well as the peak calls in a genome browser to find out if you agree with the peaks calling software Optional: remove reads mapping to the ENCODE and 1000 Genomes blacklisted regions 22
23 References Previous Hot Topics Quality Control and Mapping Reads SOPs Reviews and benchmark papers: ChIP-seq: advantages and challenges of a maturing technology (Oct 09) ( Computation for ChIP-seq and RNA-seq studies (Nov 09) ( Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLoS Comput. Biol A computational pipeline for comparative ChIP-seq analyses. Nat. Protoc ChIP-seq guidelines and practices of the ENCODE and modencode consortia. Genome Res Quality control and strand cross-correlation MACS: Model-based Analysis of ChIP-Seq (MACS). Genome Biol Using MACS to identify peaks from ChIP-Seq data. Curr Protoc Bioinformatics
ChIP- seq Analysis. BaRC Hot Topics - Feb 24 th 2015 BioinformaBcs and Research CompuBng Whitehead InsBtute. hgp://barc.wi.mit.
 ChIP- seq Analysis BaRC Hot Topics - Feb 24 th 2015 BioinformaBcs and Research CompuBng Whitehead InsBtute hgp://barc.wi.mit.edu/hot_topics/ Before we start: 1. Log into tak (step 0 on the exercises) 2.
ChIP- seq Analysis BaRC Hot Topics - Feb 24 th 2015 BioinformaBcs and Research CompuBng Whitehead InsBtute hgp://barc.wi.mit.edu/hot_topics/ Before we start: 1. Log into tak (step 0 on the exercises) 2.
ChIP-seq Analysis. BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute.
 ChIP-seq Analysis BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
ChIP-seq Analysis BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
ChIP-seq Analysis. BaRC Hot Topics - Feb 23 th 2016 Bioinformatics and Research Computing Whitehead Institute.
 ChIP-seq Analysis BaRC Hot Topics - Feb 23 th 2016 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
ChIP-seq Analysis BaRC Hot Topics - Feb 23 th 2016 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Practical 4: ChIP-seq Peak calling
 Practical 4: ChIP-seq Peak calling Shamith Samarajiiwa, Dora Bihary September 2017 Contents 1 Calling ChIP-seq peaks using MACS2 1 1.1 Assess the quality of the aligned datasets..................................
Practical 4: ChIP-seq Peak calling Shamith Samarajiiwa, Dora Bihary September 2017 Contents 1 Calling ChIP-seq peaks using MACS2 1 1.1 Assess the quality of the aligned datasets..................................
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
From genomic regions to biology
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
ChIP-seq practical: peak detection and peak annotation. Mali Salmon-Divon Remco Loos Myrto Kostadima
 ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Identifying ChIP-seq enrichment using MACS
 Identifying ChIP-seq enrichment using MACS Jianxing Feng 1,3, Tao Liu 2,3, Bo Qin 1, Yong Zhang 1 & Xiaole Shirley Liu 2 1 Department of Bioinformatics, School of Life Sciences and Technology, Tongji University,
Identifying ChIP-seq enrichment using MACS Jianxing Feng 1,3, Tao Liu 2,3, Bo Qin 1, Yong Zhang 1 & Xiaole Shirley Liu 2 1 Department of Bioinformatics, School of Life Sciences and Technology, Tongji University,
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
!"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468,
#$*)+,-./).010#,23+3,3034566,&((46,7$+-./&((468,") !"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468, 9"(1(02)1+(',:.;.4(*.',?9@A,!."2.4B.'#A,C(;.
!"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468, 9"(1(02)1+(',:.;.4(*.',?9@A,!."2.4B.'#A,C(;.
User's guide to ChIP-Seq applications: command-line usage and option summary
 User's guide to ChIP-Seq applications: command-line usage and option summary 1. Basics about the ChIP-Seq Tools The ChIP-Seq software provides a set of tools performing common genome-wide ChIPseq analysis
User's guide to ChIP-Seq applications: command-line usage and option summary 1. Basics about the ChIP-Seq Tools The ChIP-Seq software provides a set of tools performing common genome-wide ChIPseq analysis
ChIP-Seq Tutorial on Galaxy
 1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
Useful software utilities for computational genomics. Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017
 Useful software utilities for computational genomics Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017 Overview Search and download genomic datasets: GEOquery, GEOsearch and GEOmetadb,
Useful software utilities for computational genomics Shamith Samarajiwa CRUK Autumn School in Bioinformatics September 2017 Overview Search and download genomic datasets: GEOquery, GEOsearch and GEOmetadb,
Genomic Analysis with Genome Browsers.
 Genomic Analysis with Genome Browsers http://barc.wi.mit.edu/hot_topics/ 1 Outline Genome browsers overview UCSC Genome Browser Navigating: View your list of regions in the browser Available tracks (eg.
Genomic Analysis with Genome Browsers http://barc.wi.mit.edu/hot_topics/ 1 Outline Genome browsers overview UCSC Genome Browser Navigating: View your list of regions in the browser Available tracks (eg.
Today's outline. Resources. Genome browser components. Genome browsers: Discovering biology through genomics. Genome browser tutorial materials
 Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
Easy visualization of the read coverage using the CoverageView package
 Easy visualization of the read coverage using the CoverageView package Ernesto Lowy European Bioinformatics Institute EMBL June 13, 2018 > options(width=40) > library(coverageview) 1 Introduction This
Easy visualization of the read coverage using the CoverageView package Ernesto Lowy European Bioinformatics Institute EMBL June 13, 2018 > options(width=40) > library(coverageview) 1 Introduction This
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq Analysis Practical
 ChIP-seq Analysis Practical Vladimir Teif (vteif@essex.ac.uk) An updated version of this document will be available at http://generegulation.info/index.php/teaching In this practical we will learn how
ChIP-seq Analysis Practical Vladimir Teif (vteif@essex.ac.uk) An updated version of this document will be available at http://generegulation.info/index.php/teaching In this practical we will learn how
ASAP - Allele-specific alignment pipeline
 ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
Agilent Genomic Workbench Lite Edition 6.5
 Agilent Genomic Workbench Lite Edition 6.5 SureSelect Quality Analyzer User Guide For Research Use Only. Not for use in diagnostic procedures. Agilent Technologies Notices Agilent Technologies, Inc. 2010
Agilent Genomic Workbench Lite Edition 6.5 SureSelect Quality Analyzer User Guide For Research Use Only. Not for use in diagnostic procedures. Agilent Technologies Notices Agilent Technologies, Inc. 2010
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
A short Introduction to UCSC Genome Browser
 A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
de.nbi and its Galaxy interface for RNA-Seq
 de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq
 TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
TECH NOTE Improving the Sensitivity of Ultra Low Input mrna Seq SMART Seq v4 Ultra Low Input RNA Kit for Sequencing Powered by SMART and LNA technologies: Locked nucleic acid technology significantly improves
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
Introduction to Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
Bioinformatics I. Teaching assistant(s): Eudes Barbosa Markus List
: Eudes Barbosa Markus List") Bioinformatics I Lecturer: Jan Baumbach Teaching assistant(s): Eudes Barbosa Markus List Question How can we study protein/dna binding events on a genome-wide scale? 2 Outline Short outline/intro to ChIP-Sequencing
Bioinformatics I Lecturer: Jan Baumbach Teaching assistant(s): Eudes Barbosa Markus List Question How can we study protein/dna binding events on a genome-wide scale? 2 Outline Short outline/intro to ChIP-Sequencing
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1
 EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
ChIP-Seq data analysis workshop
 ChIP-Seq data analysis workshop Exercise 1. ChIP-Seq peak calling 1. Using Putty (Windows) or Terminal (Mac) to connect to your assigned computer. Create a directory /workdir/myuserid (replace myuserid
ChIP-Seq data analysis workshop Exercise 1. ChIP-Seq peak calling 1. Using Putty (Windows) or Terminal (Mac) to connect to your assigned computer. Create a directory /workdir/myuserid (replace myuserid
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Analysis of ChIP-seq Data with mosaics Package: MOSAiCS and MOSAiCS-HMM
 Analysis of ChIP-seq Data with mosaics Package: MOSAiCS and MOSAiCS-HMM Dongjun Chung 1, Pei Fen Kuan 2, Rene Welch 3, and Sündüz Keleş 3,4 1 Department of Public Health Sciences, Medical University of
Analysis of ChIP-seq Data with mosaics Package: MOSAiCS and MOSAiCS-HMM Dongjun Chung 1, Pei Fen Kuan 2, Rene Welch 3, and Sündüz Keleş 3,4 1 Department of Public Health Sciences, Medical University of
Identiyfing splice junctions from RNA-Seq data
 Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Introduction to Galaxy
 Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
modencode Galaxy: Uniform ChIP-Seq Processing Tools for modencode and ENCODE Data
 modencode Galaxy: Uniform ChIP-Seq Processing Tools for modencode and ENCODE Data Quang M Trinh Ontario Institute for Cancer Research qtrinh@oicr.on.ca Outline Model Organism ENCyclopedia Of DNA Elements
modencode Galaxy: Uniform ChIP-Seq Processing Tools for modencode and ENCODE Data Quang M Trinh Ontario Institute for Cancer Research qtrinh@oicr.on.ca Outline Model Organism ENCyclopedia Of DNA Elements
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection
 CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Advanced UCSC Browser Functions
 Advanced UCSC Browser Functions Dr. Thomas Randall tarandal@email.unc.edu bioinformatics.unc.edu UCSC Browser: genome.ucsc.edu Overview Custom Tracks adding your own datasets Utilities custom tools for
Advanced UCSC Browser Functions Dr. Thomas Randall tarandal@email.unc.edu bioinformatics.unc.edu UCSC Browser: genome.ucsc.edu Overview Custom Tracks adding your own datasets Utilities custom tools for
Automated Bioinformatics Analysis System on Chip ABASOC. version 1.1
 Automated Bioinformatics Analysis System on Chip ABASOC version 1.1 Phillip Winston Miller, Priyam Patel, Daniel L. Johnson, PhD. University of Tennessee Health Science Center Office of Research Molecular
Automated Bioinformatics Analysis System on Chip ABASOC version 1.1 Phillip Winston Miller, Priyam Patel, Daniel L. Johnson, PhD. University of Tennessee Health Science Center Office of Research Molecular
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Triform: peak finding in ChIP-Seq enrichment profiles for transcription factors
 Triform: peak finding in ChIP-Seq enrichment profiles for transcription factors Karl Kornacker * and Tony Håndstad October 30, 2018 A guide for using the Triform algorithm to predict transcription factor
Triform: peak finding in ChIP-Seq enrichment profiles for transcription factors Karl Kornacker * and Tony Håndstad October 30, 2018 A guide for using the Triform algorithm to predict transcription factor
TP RNA-seq : Differential expression analysis
 TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
PICS: Probabilistic Inference for ChIP-Seq
 PICS: Probabilistic Inference for ChIP-Seq Xuekui Zhang * and Raphael Gottardo, Arnaud Droit and Renan Sauteraud April 30, 2018 A step-by-step guide in the analysis of ChIP-Seq data using the PICS package
PICS: Probabilistic Inference for ChIP-Seq Xuekui Zhang * and Raphael Gottardo, Arnaud Droit and Renan Sauteraud April 30, 2018 A step-by-step guide in the analysis of ChIP-Seq data using the PICS package
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
Exercise 1 Review. --outfiltermismatchnmax : max number of mismatch (Default 10) --outreadsunmapped fastx: output unmapped reads
 --outreadsunmapped fastx: output unmapped reads") Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Pre-processing and quality control of sequence data. Barbera van Schaik KEBB - Bioinformatics Laboratory
 Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
RNA- SeQC Documentation
 RNA- SeQC Documentation Description: Author: Calculates metrics on aligned RNA-seq data. David S. DeLuca (Broad Institute), gp-help@broadinstitute.org Summary This module calculates standard RNA-seq related
RNA- SeQC Documentation Description: Author: Calculates metrics on aligned RNA-seq data. David S. DeLuca (Broad Institute), gp-help@broadinstitute.org Summary This module calculates standard RNA-seq related
Exome sequencing. Jong Kyoung Kim
 Exome sequencing Jong Kyoung Kim Genome Analysis Toolkit The GATK is the industry standard for identifying SNPs and indels in germline DNA and RNAseq data. Its scope is now expanding to include somatic
Exome sequencing Jong Kyoung Kim Genome Analysis Toolkit The GATK is the industry standard for identifying SNPs and indels in germline DNA and RNAseq data. Its scope is now expanding to include somatic
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Gene Expression Data Analysis. Qin Ma, Ph.D. December 10, 2017
 1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
SICER 1.1. If you use SICER to analyze your data in a published work, please cite the above paper in the main text of your publication.
 SICER 1.1 1. Introduction For details description of the algorithm, please see A clustering approach for identification of enriched domains from histone modification ChIP-Seq data Chongzhi Zang, Dustin
SICER 1.1 1. Introduction For details description of the algorithm, please see A clustering approach for identification of enriched domains from histone modification ChIP-Seq data Chongzhi Zang, Dustin
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Sequence Preprocessing: A perspective
 Sequence Preprocessing: A perspective Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu Why Preprocess reads We have found that aggressively cleaning and processing
Sequence Preprocessing: A perspective Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu Why Preprocess reads We have found that aggressively cleaning and processing
Nature Biotechnology: doi: /nbt Supplementary Figure 1
 Supplementary Figure 1 Detailed schematic representation of SuRE methodology. See Methods for detailed description. a. Size-selected and A-tailed random fragments ( queries ) of the human genome are inserted
Supplementary Figure 1 Detailed schematic representation of SuRE methodology. See Methods for detailed description. a. Size-selected and A-tailed random fragments ( queries ) of the human genome are inserted
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
Run Setup and Bioinformatic Analysis. Accel-NGS 2S MID Indexing Kits
 Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
The ChIP-seq quality Control package ChIC: A short introduction
 The ChIP-seq quality Control package ChIC: A short introduction April 30, 2018 Abstract The ChIP-seq quality Control package (ChIC) provides functions and data structures to assess the quality of ChIP-seq
The ChIP-seq quality Control package ChIC: A short introduction April 30, 2018 Abstract The ChIP-seq quality Control package (ChIC) provides functions and data structures to assess the quality of ChIP-seq
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
How To: Run the ENCODE histone ChIP- seq analysis pipeline on DNAnexus
 How To: Run the ENCODE histone ChIP- seq analysis pipeline on DNAnexus Overview: In this exercise, we will run the ENCODE Uniform Processing ChIP- seq Pipeline on a small test dataset containing reads
How To: Run the ENCODE histone ChIP- seq analysis pipeline on DNAnexus Overview: In this exercise, we will run the ENCODE Uniform Processing ChIP- seq Pipeline on a small test dataset containing reads
Accessible, Transparent and Reproducible Analysis with Galaxy
 Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
Rsubread package: high-performance read alignment, quantification and mutation discovery
 Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units
 GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
GPUBwa -Parallelization of Burrows Wheeler Aligner using Graphical Processing Units Abstract A very popular discipline in bioinformatics is Next-Generation Sequencing (NGS) or DNA sequencing. It specifies
Introduction to GE Microarray data analysis Practical Course MolBio 2012
 Introduction to GE Microarray data analysis Practical Course MolBio 2012 Claudia Pommerenke Nov-2012 Transkriptomanalyselabor TAL Microarray and Deep Sequencing Core Facility Göttingen University Medical
Introduction to GE Microarray data analysis Practical Course MolBio 2012 Claudia Pommerenke Nov-2012 Transkriptomanalyselabor TAL Microarray and Deep Sequencing Core Facility Göttingen University Medical
Welcome to GenomeView 101!
 Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Analyzing ChIP-Seq Data at the Command Line
 Analyzing ChIP-Seq Data at the Command Line Quick UNIX Introduction: UNIX is an operating system like OSX or Windows. The interface between you and the UNIX OS is called the shell. There are a few flavors
Analyzing ChIP-Seq Data at the Command Line Quick UNIX Introduction: UNIX is an operating system like OSX or Windows. The interface between you and the UNIX OS is called the shell. There are a few flavors
DNase I Seq data Analysis Strategy. Dragon Star 2013 QianQin 同济大学
 DNase I Seq data Analysis Strategy Dragon Star 2013 QianQin 同济大学 Workflow Mapping(BWA/Bow8e) QC Reads filtering and format(samtools /Picard) qrqc, FastQC 1. Sampling down by mappable reads 2. Scale mappable
DNase I Seq data Analysis Strategy Dragon Star 2013 QianQin 同济大学 Workflow Mapping(BWA/Bow8e) QC Reads filtering and format(samtools /Picard) qrqc, FastQC 1. Sampling down by mappable reads 2. Scale mappable
Next Generation Sequence Alignment on the BRC Cluster. Steve Newhouse 22 July 2010
 Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
NGS Analyses with Galaxy
 1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
Using Galaxy to provide a NGS Analysis Platform GTC s NGS & Bioinformatics Summit Europe October 7-8, 2013 in Berlin, Germany.
 Using Galaxy to provide a NGS Analysis Platform GTC s NGS & Bioinformatics Summit Europe October 7-8, 2013 in Berlin, Germany. (public version) Hans-Rudolf Hotz ( hrh@fmi.ch ) Friedrich Miescher Institute
Using Galaxy to provide a NGS Analysis Platform GTC s NGS & Bioinformatics Summit Europe October 7-8, 2013 in Berlin, Germany. (public version) Hans-Rudolf Hotz ( hrh@fmi.ch ) Friedrich Miescher Institute
Aligners. J Fass 23 August 2017
 Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Aligners J Fass 23 August 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-08-23
Next-Generation Sequencing applied to adna
 Next-Generation Sequencing applied to adna Hands-on session June 13, 2014 Ludovic Orlando - Lorlando@snm.ku.dk Mikkel Schubert - MSchubert@snm.ku.dk Aurélien Ginolhac - AGinolhac@snm.ku.dk Hákon Jónsson
Next-Generation Sequencing applied to adna Hands-on session June 13, 2014 Ludovic Orlando - Lorlando@snm.ku.dk Mikkel Schubert - MSchubert@snm.ku.dk Aurélien Ginolhac - AGinolhac@snm.ku.dk Hákon Jónsson