TP RNA-seq : Differential expression analysis
|
|
|
- Quentin Casey
- 5 years ago
- Views:
Transcription
1 TP RNA-seq : Differential expression analysis
2 Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
3 Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 3
4 Input data 8 fastq files : RNA-seq paired-end Illumina 2 x 75 pb Ex : Hct116_rep1_R1.fastq cell line replicate pair 1 fasta file : reference genome sequence chr22.fasta 1 gtf file : reference genome annotation chr22.gtf 4

![Then click on «[FORMATION] Input Data» and select the folder](/docs-images/83/87102078/images/5-1.jpg "«Roscoff», then validate by clicking on «GO», at the bottom of")
5 Data import Go to the web page : Go to «Shared Data» and click on «Data Librairies» Then click on «[FORMATION] Input Data» and select the folder «Roscoff», then validate by clicking on «GO», at the bottom of the page 5


6 Data import Return at the home page by clicking on «Analyze data» Rename the history, click on «Unnamed history» then type a new name and press enter 6
7 Steps of analysis Quality control Mapping Quantification Differential analysis Formation NGS & Cancer - Analyses RNA-Seq novembre 2014
8 Steps of analysis Quality control Mapping Quantification Differential analysis Formation NGS & Cancer - Analyses RNA-Seq novembre 2014
9 Steps of analysis Quality control FastQC FASTQ Trimmer Mapping Quantification Differential analysis Formation NGS & Cancer - Analyses RNA-Seq novembre 2014


10 Quality control In the left panel, type «fastqc» in the search tool, then click on «FastQC:Read QC» Select «Gm12878_rep1_R1.fastq» sample and click on «Execute» 10



11 Quality control Run the tool again by clicking on the rerun icon, and select «Gm12878_rep1_R2.fastq» sample To view the results, click on the eye on the dataset 11

12 Quality control Encoding quality 12
13 Quality control The end of the sequence is generally of poor quality Trimming 13


14 Fastq trimming Type «fastq trim» on the search tool, then select the «FASTQ Quality Trimmer» tool. Galaxy is don't aware of the quality of the fastq The datatype of the fastq need to be changed manually 14


15 Change datatype To change the dataype, click on the pencil icon on the dataset Then click on «Dataype», search the fastqsanger format and validate by clicking on «Save» Repeat this operation for all the fastq 15
16 Fastq trimming Use the «FASTQ Quality Trimmer» tool on Gm12878_rep1_R1.fastq 16

17 Fastq trimming Repeat this operation for Gm12878_rep1_R2.fastq 17


18 Fastq trimming Rename the outputs of fastq trimming, click on the pencil icon, type a new name and don't forget to save. 18

19 Quality control You can check the result of trimming by running «FastQC:Read QC» on the trimmed fastq. 19

20 Quality control Before trimming After trimming 20
21 Steps of analysis Quality control Mapping Tophat2 Quantification Differential analysis Formation NGS & Cancer - Analyses RNA-Seq novembre 2014
22 Mapping Which mapper? (Fonseca N A et al. Bioinformatics 2012;28: ) 22
23 Mapping Tophat2 : - Use Bowtie2 - splice aware - optimised for 75 pb reads or longer - several options : not all include in galaxy - different mode (ex : tophat-fusion) - tophat+tophat citations - alternatives : STAR, RUM, Mapsplice (Trapnell C et al. Bioinformatics 2009 ;25: ) 23


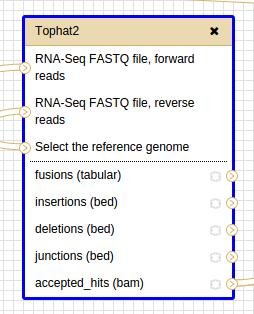
24 Mapping In the left panel in the section «NGS : RNA-seq Fusion», click on «Tophat2» 24

25 Mapping You can run an other job on the dataset even if is not terminated We use the tool «flagstat» to have statistics on the mapping 25


26 Mapping Tophat outputs : - bed files : coordinates of splice junctions, insertions or deletions - bam file : accepted_hits mapped reads BAM = binary alignment format Bam-to-Sam SAM = sequence alignment format 26
27 Mapping Header SAM file ID:TopHat SO:coordinate LN: VN: CL:/bioinfo/local/build/tophat /tophat -p 4 -r 200 [ ] Read 616L7AAXX_HWUSI:2:41:19518: chr M = GTGCCTGGCTGACTTATTGGCATTTCTAACAGAGAAGAAGAGAAAGTAAGCAACTTGGAAAACATTTTTGAGGAT GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGBGGGGEGGGGFGGGFGGGGFFGFGGGEFDGGGGGGGGFAGEGG AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:75 YT:Z:UU XP:Z:chr M 213 Mandatory fields 616L7AAXX_HWUSI:2:41:19518: Read sequence AGAACTCCCGTGAGACTGAAGGTAGGCAGTGAAGCAAATGTTTGCATTCTTGTGTGGCTCTGATTAGCATCAGGA Read quality DFFDF=FEFFGGGGGGFGGGGGGGGEGGGFGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG Optionnal fields chr AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:75 50 YT:Z:UU 75M = XP:Z:chr M NH:i:1 NH:i:1 27


28 Mapping SAM file : Read 616L7AAXX_HWUSI:2:41:19518: chr M = GTGCCTGGCTGACTTATTGGCATTTCTAACAGAGAAGAAGAGAAAGTAAGCAACTTGGAAAACATTTTTGAGGAT GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGBGGGGEGGGGFGGGFGGGGFFGFGGGEFDGGGGGGGGFAGEGG AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:75 YT:Z:UU XP:Z:chr M L7AAXX_HWUSI:2:41:19518: chr M = AGAACTCCCGTGAGACTGAAGGTAGGCAGTGAAGCAAATGTTTGCATTCTTGTGTGGCTCTGATTAGCATCAGGA DFFDF=FEFFGGGGGGFGGGGGGGGEGGGFGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:75 YT:Z:UU XP:Z:chr M -213 NH:i:1 NH:i:1 Flags Explain SAM flags : 28



29 Mapping Flagstat output : Rename the «accepted_hits» dataset 29


30 Mapping Visualization with IGV - download the bam file : click on the disk icon - download the bam_index : click on the arrow then select «Download bam_index» 30


31 Mapping - Open IGV and click on «File», then «Load from file», select the bam file, click on open 31

32 Mapping - To view alignment, you can specify a gene of your choice, here we use LGALS1 as example 32
33 Steps of analysis Quality control Mapping Quantification HTSeq-count Differential analysis 33

34 Quantification Search the tool «htseq-count», change the stranded option to «No» and click on «Execute» 34
35 Quantification HTSeq-count : Counting reads in features - input : BAM file GTF file reference annotation - feature type and ID Attribute : chr20 protein_coding gene_name "ZNF366"; chr20 protein_coding gene_name "ZNF366"; chr20 protein_coding gene_name "ZNF366"; chr20 protein_coding gene_name "ZNF366"; chr20 protein_coding gene_name "ZNF366"; exon gene_id "ENSBTAG "; transcript_id "ENSBTAT "; CDS gene_id "ENSBTAG "; transcript_id "ENSBTAT "; exon gene_id "ENSBTAG "; transcript_id "ENSBTAT "; CDS gene_id "ENSBTAG "; transcript_id "ENSBTAT "; exon gene_id "ENSBTAG "; transcript_id "ENSBTAT "; - counting mode : union 35

36 Quantification 36
37 Steps of analysis Quality control Mapping Workflow Quantification Differential analysis 37

38 Workflow Rename the htseq-count output Click on the wheel of History panel, then click on «Extract Workflow» 38


39 Workflow Uncheck the unusued datasets, then click on «Create Workflow» 39

40 Workflow Click on «Workflow» on the top menu Click on the Workflow that have been created, and select «Edit» 40

41 Workflow You can view the schema of the Workflow, move the datasets to organize the workflow like you want 41





42 Workflow Change the name of the output files 42


43 Workflow Click on the wheel of the Workflow panel and select «Save» then «Run» Now, we can run the workflow on the 3 others samples : Gm12878_rep2, Hct116_rep1 and Hct116_rep2 43

44 Workflow Click on the pages to enable list Select the R1 of sample Select the R2 of sample 44
45 Steps of analysis Quality control Mapping Quantification Differential analysis DESeq 45
46 Differential analysis Search and select the «DESeq» tool 46
47 Differential analysis 47
48 Differential analysis DESeq : differential expression analysis for sequence count data - at gene level transcripts, exons levels : DEXSeq - comparison of 2 conditions complex designs (ex : Time-series) : DESeq2 - alernatives : edger, limma DESeq manual : /DESeq/inst/doc/DESeq.pdf 48
49 Differential analysis Steps of analysis with DESeq : - Normalization of expression : comparable expression - Variance estimation : by the negative binomial distribution - Inference : calling of differential expression Choice of parameters : - mode : explication of dispersion - method : impact of design - type : fonction to estimate dispersion 49
50 Differential analysis Outputs of deseq : - html file : figures to understand and verify the experiment - 2 tabular files : results for significantly over-expressed genes and sub-expressed genes. id = gene identification basemean = normalized mean counts foldchange = mean ratios pval = p value padj = adjusted p value 50

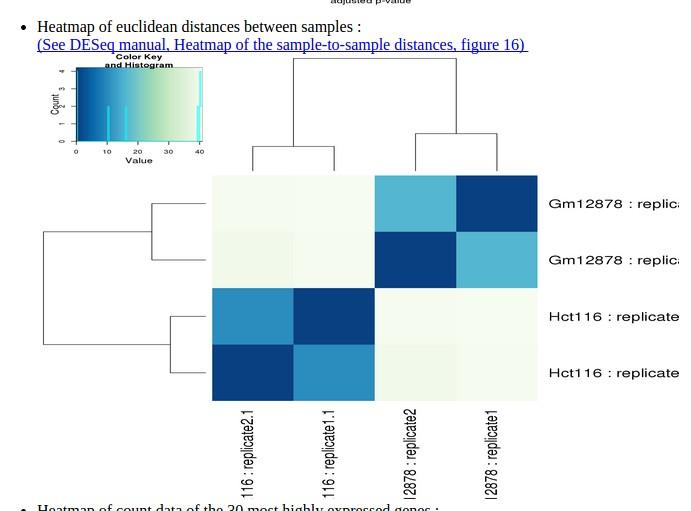
51 Differential analysis 51
52 Differential analysis 52
53 Differential analysis Differentially expressed genes Low counts Not differentially expressed genes (Ex : on the entire genome) 53
54 Differential analysis 54

55 Differential analysis 55
56 Differential analysis - Red line : estimated dispersion - Black dots : observed dispersion 56
57 Differential analysis Overexpression cond1 MA-plot : Mean Average Plot Overexpression Cond2 Significant genes Not significant genes FoldChange = 1 Genes expression 57
58 Differential analysis Volcano Plot -/+ Inf : not expressed genes in one condition Significance Significant genes Not significant genes Overexpression cond2 Overexpression cond1 58
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
File Formats: SAM, BAM, and CRAM. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
de.nbi and its Galaxy interface for RNA-Seq
 de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
Services Performed. The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples.
 Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
Goal: Learn how to use various tool to extract information from RNAseq reads. 4.1 Mapping RNAseq Reads to a Genome Assembly
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2014 UNIVERSITY OF KENTUCKY AGTC Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Using the Galaxy Local Bioinformatics Cloud at CARC
 Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University
Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
INF-BIO5121/ Oct 7, Analyzing mirna data using Lifeportal PRACTICALS
 INF-BIO5121/9121 - Oct 7, 2014 Analyzing mirna data using Lifeportal PRACTICALS In this experiment we have mirna data from the livers of baboons (Papio Hamadryas) before and after they are given a high
INF-BIO5121/9121 - Oct 7, 2014 Analyzing mirna data using Lifeportal PRACTICALS In this experiment we have mirna data from the livers of baboons (Papio Hamadryas) before and after they are given a high
NGS : reads quality control
 NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv
 Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Tutorial. RNA-Seq Analysis of Breast Cancer Data. Sample to Insight. November 21, 2017
 RNA-Seq Analysis of Breast Cancer Data November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com AdvancedGenomicsSupport@qiagen.com
RNA-Seq Analysis of Breast Cancer Data November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com AdvancedGenomicsSupport@qiagen.com
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
The QoRTs Analysis Pipeline Example Walkthrough
 The QoRTs Analysis Pipeline Example Walkthrough Stephen Hartley National Human Genome Research Institute National Institutes of Health October 31, 2017 QoRTs v1.0.1 JunctionSeq v1.9.0 Contents 1 Overview
The QoRTs Analysis Pipeline Example Walkthrough Stephen Hartley National Human Genome Research Institute National Institutes of Health October 31, 2017 QoRTs v1.0.1 JunctionSeq v1.9.0 Contents 1 Overview
NGS Analyses with Galaxy
 1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Gene expression estimation
 Gene expression estimation") mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
JunctionSeq Package User Manual
 JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health March 30, 2017 JunctionSeq v1.5.4 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health March 30, 2017 JunctionSeq v1.5.4 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
Briefly: Bioinformatics File Formats. J Fass September 2018
 Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
Analysis of ChIP-seq data
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Differential Expression
 Differential Expression Data In this practical, as before, we will work with RNA-Seq data from Arabidopsis seeds that matured at standard temperature (ST, 22 C day/18 C night) or at high temperature (HT,
Differential Expression Data In this practical, as before, we will work with RNA-Seq data from Arabidopsis seeds that matured at standard temperature (ST, 22 C day/18 C night) or at high temperature (HT,
!"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468,
#$*)+,-./).010#,23+3,3034566,&((46,7$+-./&((468,") !"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468, 9"(1(02)1+(',:.;.4(*.',?9@A,!."2.4B.'#A,C(;.
!"#$%&$'()#$*)+,-./).01"0#,23+3,303456"6,&((46,7$+-./&((468, 9"(1(02)1+(',:.;.4(*.',?9@A,!."2.4B.'#A,C(;.
Exercise 1 Review. --outfiltermismatchnmax : max number of mismatch (Default 10) --outreadsunmapped fastx: output unmapped reads
 --outreadsunmapped fastx: output unmapped reads") Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
Exercise 1 Review Setting parameters STAR --quantmode GeneCounts --genomedir genomedb -- runthreadn 2 --outfiltermismatchnmax 2 --readfilesin WTa.fastq.gz --readfilescommand zcat --outfilenameprefix WTa
Reference guided RNA-seq data analysis using BioHPC Lab computers
 Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
version /1/2011 Source code Linux x86_64 binary Mac OS X x86_64 binary
 Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
Cufflinks RNA-Seq analysis tools - Getting Started 1 of 6 14.07.2011 09:42 Cufflinks Transcript assembly, differential expression, and differential regulation for RNA-Seq Site Map Home Getting started
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, September/October 2018 Friederike Dündar with Luce Skrabanek & Paul Zumbo Day 3: Counting reads
Exercises: Analysing RNA-Seq data
 Exercises: Analysing RNA-Seq data Version 2018-03 Exercises: Analysing RNA-Seq data 2 Licence This manual is 2011-18, Simon Andrews, Laura Biggins. This manual is distributed under the creative commons
Exercises: Analysing RNA-Seq data Version 2018-03 Exercises: Analysing RNA-Seq data 2 Licence This manual is 2011-18, Simon Andrews, Laura Biggins. This manual is distributed under the creative commons
Analyzing Variant Call results using EuPathDB Galaxy, Part II
 Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day four Quantifying expression Intro to R Differential expression
The QoRTs Analysis Pipeline Example Walkthrough
 The QoRTs Analysis Pipeline Example Walkthrough Stephen Hartley National Human Genome Research Institute National Institutes of Health March 30, 2017 QoRTs v1.2.0 JunctionSeq v1.5.4 Contents 1 Overview
The QoRTs Analysis Pipeline Example Walkthrough Stephen Hartley National Human Genome Research Institute National Institutes of Health March 30, 2017 QoRTs v1.2.0 JunctionSeq v1.5.4 Contents 1 Overview
INTRODUCTION AUX FORMATS DE FICHIERS
 INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
11/8/2017 Trinity De novo Transcriptome Assembly Workshop trinityrnaseq/rnaseq_trinity_tuxedo_workshop Wiki GitHub
 trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
trinityrnaseq / RNASeq_Trinity_Tuxedo_Workshop Trinity De novo Transcriptome Assembly Workshop Brian Haas edited this page on Oct 17, 2015 14 revisions De novo RNA-Seq Assembly and Analysis Using Trinity
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
Accessible, Transparent and Reproducible Analysis with Galaxy
 Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
Accessible, Transparent and Reproducible Analysis with Galaxy Application of Next Generation Sequencing Technologies for Whole Transcriptome and Genome Analysis ABRF 2013 Saturday, March 2, 2013 Palm Springs,
Helpful Galaxy screencasts are available at:
 This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
Centre (CNIO). 3rd Melchor Fernández Almagro St , Madrid, Spain. s/n, Universidad de Vigo, Ourense, Spain.
. 3rd Melchor Fernández Almagro St , Madrid, Spain. s/n, Universidad de Vigo, Ourense, Spain.") O. Graña *a,b, M. Rubio-Camarillo a, F. Fdez-Riverola b, D.G. Pisano a and D. Glez-Peña b a Bioinformatics Unit, Structural Biology and BioComputing Programme, Spanish National Cancer Research Centre (CNIO).
O. Graña *a,b, M. Rubio-Camarillo a, F. Fdez-Riverola b, D.G. Pisano a and D. Glez-Peña b a Bioinformatics Unit, Structural Biology and BioComputing Programme, Spanish National Cancer Research Centre (CNIO).
JunctionSeq Package User Manual
 JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health v0.6.10 November 20, 2015 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health v0.6.10 November 20, 2015 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
Exercise 1. RNA-seq alignment and quantification. Part 1. Prepare the working directory. Part 2. Examine qualities of the RNA-seq data files
 Exercise 1. RNA-seq alignment and quantification Part 1. Prepare the working directory. 1. Connect to your assigned computer. If you do not know how, follow the instruction at http://cbsu.tc.cornell.edu/lab/doc/remote_access.pdf
Exercise 1. RNA-seq alignment and quantification Part 1. Prepare the working directory. 1. Connect to your assigned computer. If you do not know how, follow the instruction at http://cbsu.tc.cornell.edu/lab/doc/remote_access.pdf
ChIP-Seq Tutorial on Galaxy
 1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat a.tgz. Software:
 A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
A Tutorial: De novo RNA- Seq Assembly and Analysis Using Trinity and edger The following data and software resources are required for following the tutorial: Data: ftp://ftp.broad.mit.edu/pub/users/bhaas/rnaseq_workshop/rnaseq_workshop_dat
RNA-Seq data analysis software. User Guide 023UG050V0200
 RNA-Seq data analysis software User Guide 023UG050V0200 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
RNA-Seq data analysis software User Guide 023UG050V0200 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
Using Galaxy: RNA-seq
 Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Supplementary Figure 1. Fast read-mapping algorithm of BrowserGenome.
 Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Introduction to Galaxy
 Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
JunctionSeq Package User Manual
 JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health February 16, 2016 JunctionSeq v1.1.3 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
JunctionSeq Package User Manual Stephen Hartley National Human Genome Research Institute National Institutes of Health February 16, 2016 JunctionSeq v1.1.3 Contents 1 Overview 2 2 Requirements 3 2.1 Alignment.........................................
ChIP-seq practical: peak detection and peak annotation. Mali Salmon-Divon Remco Loos Myrto Kostadima
 ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
Cycle «Analyse de données de séquençage à haut-débit» Module 1/5 Analyse ADN. Sophie Gallina CNRS Evo-Eco-Paléo (EEP)
") Cycle «Analyse de données de séquençage à haut-débit» Module 1/5 Analyse ADN Sophie Gallina CNRS Evo-Eco-Paléo (EEP) (sophie.gallina@univ-lille1.fr) Module 1/5 Analyse DNA NGS Introduction Galaxy : upload
Cycle «Analyse de données de séquençage à haut-débit» Module 1/5 Analyse ADN Sophie Gallina CNRS Evo-Eco-Paléo (EEP) (sophie.gallina@univ-lille1.fr) Module 1/5 Analyse DNA NGS Introduction Galaxy : upload
From the Schnable Lab:
 From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
preparation methods and new bacterial strains. Parts of the pipeline that can be updated will be annotated in this guide.
 BacSeq Introduction The purpose of this guide is to aid current and future Whiteley Lab members and University of Texas microbiologists with bacterial RNA?Seq analysis. Once you have analyzed your data
BacSeq Introduction The purpose of this guide is to aid current and future Whiteley Lab members and University of Texas microbiologists with bacterial RNA?Seq analysis. Once you have analyzed your data
Identiyfing splice junctions from RNA-Seq data
 Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
miarma-seq: mirna-seq And RNA-Seq Multiprocess Analysis tool. mrna detection from RNA-Seq Data User s Guide
 miarma-seq: mirna-seq And RNA-Seq Multiprocess Analysis tool. mrna detection from RNA-Seq Data User s Guide Eduardo Andrés-León, Rocío Núñez-Torres and Ana M Rojas. Instituto de Biomedicina de Sevilla
miarma-seq: mirna-seq And RNA-Seq Multiprocess Analysis tool. mrna detection from RNA-Seq Data User s Guide Eduardo Andrés-León, Rocío Núñez-Torres and Ana M Rojas. Instituto de Biomedicina de Sevilla
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Gene Expression Data Analysis. Qin Ma, Ph.D. December 10, 2017
 1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
1 Gene Expression Data Analysis Qin Ma, Ph.D. December 10, 2017 2 Bioinformatics Systems biology This interdisciplinary science is about providing computational support to studies on linking the behavior
Differential gene expression analysis
 Differential gene expression analysis Overview In this exercise, we will analyze RNA-seq data to measure changes in gene expression levels between wild-type and a mutant strain of the bacterium Listeria
Differential gene expression analysis Overview In this exercise, we will analyze RNA-seq data to measure changes in gene expression levels between wild-type and a mutant strain of the bacterium Listeria
Standard output. Some of the output files can be redirected into the standard output, which may facilitate in creating the pipelines:
 Lecture 18 RNA-seq Alignment Standard output Some of the output files can be redirected into the standard output, which may facilitate in creating the pipelines: Filtering of the alignments STAR performs
Lecture 18 RNA-seq Alignment Standard output Some of the output files can be redirected into the standard output, which may facilitate in creating the pipelines: Filtering of the alignments STAR performs
Benchmarking of RNA-seq aligners
 Lecture 17 RNA-seq Alignment STAR Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Based on this analysis the most reliable
Lecture 17 RNA-seq Alignment STAR Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Benchmarking of RNA-seq aligners Based on this analysis the most reliable
RNA-Seq data analysis software. User Guide 023UG050V0100
 RNA-Seq data analysis software User Guide 023UG050V0100 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
RNA-Seq data analysis software User Guide 023UG050V0100 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
Expression Analysis with the Advanced RNA-Seq Plugin
 Expression Analysis with the Advanced RNA-Seq Plugin May 24, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Expression Analysis with the Advanced RNA-Seq Plugin May 24, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
replace my_user_id in the commands with your actual user ID
 Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
A short Introduction to UCSC Genome Browser
 A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
A short Introduction to UCSC Genome Browser Elodie Girard, Nicolas Servant Institut Curie/INSERM U900 Bioinformatics, Biostatistics, Epidemiology and computational Systems Biology of Cancer 1 Why using
Getting Started. April Strand Life Sciences, Inc All rights reserved.
 Getting Started April 2015 Strand Life Sciences, Inc. 2015. All rights reserved. Contents Aim... 3 Demo Project and User Interface... 3 Downloading Annotations... 4 Project and Experiment Creation... 6
Getting Started April 2015 Strand Life Sciences, Inc. 2015. All rights reserved. Contents Aim... 3 Demo Project and User Interface... 3 Downloading Annotations... 4 Project and Experiment Creation... 6
RNASeq2017 Course Salerno, September 27-29, 2017
 RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
RNA-Seq data analysis software. User Guide 023UG050V0210
 RNA-Seq data analysis software User Guide 023UG050V0210 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
RNA-Seq data analysis software User Guide 023UG050V0210 FOR RESEARCH USE ONLY. NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE. INFORMATION IN THIS DOCUMENT IS SUBJECT TO CHANGE WITHOUT NOTICE. Lexogen
Importing your Exeter NGS data into Galaxy:
 Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Genome 373: Mapping Short Sequence Reads III. Doug Fowler
 Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Package RNASeqR. January 8, 2019
 Type Package Package RNASeqR January 8, 2019 Title RNASeqR: RNA-Seq workflow for case-control study Version 1.1.3 Date 2018-8-7 Author Maintainer biocviews Genetics, Infrastructure,
Type Package Package RNASeqR January 8, 2019 Title RNASeqR: RNA-Seq workflow for case-control study Version 1.1.3 Date 2018-8-7 Author Maintainer biocviews Genetics, Infrastructure,
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Introduction to Cancer Genomics
 Introduction to Cancer Genomics Gene expression data analysis part I David Gfeller Computational Cancer Biology Ludwig Center for Cancer research david.gfeller@unil.ch 1 Overview 1. Basic understanding
Introduction to Cancer Genomics Gene expression data analysis part I David Gfeller Computational Cancer Biology Ludwig Center for Cancer research david.gfeller@unil.ch 1 Overview 1. Basic understanding
LFCseq: a nonparametric approach for differential expression analysis of RNA-seq data - supplementary materials
 LFCseq: a nonparametric approach for differential expression analysis of RNA-seq data - supplementary materials Bingqing Lin 1, Li-Feng Zhang 1, and Xin Chen 1 School of Biological Sciences, School of
LFCseq: a nonparametric approach for differential expression analysis of RNA-seq data - supplementary materials Bingqing Lin 1, Li-Feng Zhang 1, and Xin Chen 1 School of Biological Sciences, School of
Fusion Detection Using QIAseq RNAscan Panels
 Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com