NGS Analysis Using Galaxy
|
|
|
- Samson Cross
- 6 years ago
- Views:
Transcription
1 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises 1

2 UCR Galaxy homepage (
3 SNP-seq Analysis dataset Data source: SRR sample from experiment published by Kaufman et al (2012, GSE20176) Understand the target molecular mechanisms underlying AP1 func;on Fastq File: hxp://biocluster.ucr.edu/~rkaundal/galaxy_workshop/snpseq/srr fastq TAIR10 Genome: hxp://biocluster.ucr.edu/~rkaundal/galaxy_workshop/snpseq/tair10chr.fasta
 Filter reads based on quality")
 SAM to BAM conversion Variant calling Mpileup")
4 SNP-seq pipeline Upload data and QC Sequence (fastq file) Reference genome (fasta file) Filter reads based on quality scores Alignment with BWA Align with BWA (Burrows Wheeler Aligner) SAM to BAM conversion Variant calling Mpileup Bcftools view

5 Upload data Go to "Get Data, click open "Upload File from your computer. Then specify the following list of URLs in URL/Text box 5


6 Convert Fastq file to sanger format Select NGS: QC and manipulation and Fastq groomer The FASTQ Groomer tool is used to verify and convert between the known FASTQ variants. After grooming, the user is presented with a valid FASTQ format that is accepted by all downstream analysis tools. 6

7 Features available in history panel View, Edit, Delete file Size of the file Save the file Repeat the analysis

8 FASTQ Summary Statistics To understand the quality proper;es of the reads, one can run the FASTQ Summary Sta;s;cs tool from NGS: QC and manipula;on. 8

9 FASTQ Quality control } To understand the quality proper;es of the reads, one can also run the FASTQC: Read QC reports from NGS: QC and manipula;on. 9

10 Quality control output 10



11 Quality control reports Per base sequence quality Per base sequence content Per sequence GC content 11

12 Quality filter This tool filters reads based on quality scores. NGS: QC and manipula;on - > Generic FASTQ manipula;on- >Filter FASTQ reads by quality score and length 12
 Go to \"NGS: Mapping and click on \"Map with BWA.")
13 Alignment with BWA BWA is a fast and accurate short read aligner that allows mismatches and indels (Link) Go to "NGS: Mapping and click on "Map with BWA. (Selngs) 13

14 SAM to BAM format conversion Produce an indexed BAM file based on a sorted input SAM file. Go to "NGS: SAM Tools, then click open "SAM- to- BAM. 14

15 Variants calling with Mpileup SNP and INDEL caller to Generate BCF (Binary Variant Format) for one or mul;ple BAM files Go to "NGS: SAM Tools, then click open "MPileup SNP and indel caller. 15
16 Bcftools view Converts BCF format to VCF format. Go to "NGS: SAM Tools, then click open "bcftools view 16

17 Rename history
18 Extract workflow from history
19 Exercise 2: RNA-seq Analysis Data source: RNA- seq experiment SRA Four Samples: Samples Factors Fastq AP3_f14 AP3 AP3_f14 AP3 T1_f14 TRL T1_f14 TRL 19




20 RNA-seq Analysis workflow Input data Upload the fastq files, reference genome and GTF file Use fastq groomer to groom the fastq sequences Alignment TopHat for alignment Results in inser;ons, dele;ons, splice junctions and accepted hits Use Cuffdiff for differen;al expression tes;ng Differen;al expression tes;ng Finds significant changes in gene expression, splicing and promoter use
 Upload TAIR10.GTF with URL, specify the format \"gtf (http://biocluster.ucr.")
21 Upload Data Upload four fastq files with URL Upload tair10chr.fa with URL ( Upload TAIR10.GTF with URL, specify the format "gtf ( 21

22 Fastq Groomer Select NGS: QC and manipula;on and Fastq groomer Run Fastq Groomer for all the 4 fastq sequences. 22
23 RNA-seq Analysis

24 Alignment with TopHat TopHat is a fast splice junction mapper for RNA- Seq reads, it can iden;fy splice junctions between exons. Go to "NGS RNA analysis, click open "Tophat for illumina Similarly repeat this process for all the 4 fastq groomed sequences 24

25 Find Significant Changes Cuffdiff find significant changes in transcript expression. Go to "NGS RNA analysis, click open "Cuffdiff 25
26 Cuffdiff Output TSS... files report on Transcrip;on Start Sites splicing... report on splicing CDS... track coding region expression transcript... track transcripts gene... rolls up the transcripts into their genes gene/transcript FPKM tracking: gives informa;on about the gene/ transcript (length, nearest ref id, TSS, etc) and the confidence intervals for FPKM for each condition. gene/transcript differen;al expression tes;ng: gives the expression change between groups, a status of whether there was enough data for that value to be accurate (OK is good, FAIL and NOTEST are bad. LOWDATA is somewhere in between). Finally, it gives a p- value. see more details Link 26

27 Find Significant Changes Cuffdiff find significant changes in transcript expression 27
28 Output Files from Galaxy SNP- seq Save Bam file BWA generated Save.bai file (index of BAM) BWA generated Save vcf file Samtools mpileup generated Already saved them at: RNA- seq Save four Bam files and four.bai files Already saved them at: 28

29 Downloading bam index file (.bai)
30 Outline What is Galaxy Galaxy for Bioinforma;cians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualiza;on and Explora;on Using IGV 30

31 Why IGV IGV is an integrated visualiza;on tool of large data types View large dataset easily Faster naviga;on on browsing Run it locally on your computer Easy to use interface

32 IGV download 32


33 IGV interface Specify genomic region Zoom in Ruler Tracks

34 Load data Select genome: Click the genome drop- down list in the toolbar and select the genome Select chromosome 34

35 Load from URL, file, server Load data files 35
36 Toolbar Genome drop- down box: loads a genome Chromosome drop- down box: zooms to a chromosome Search box: Displays the chromosome loca;on being shown. To scroll to a different loca;on, enter the gene name, locus or track name and click Go. Whole genome view: Zooms to whole genome view. Define a region: Defines a region of interest on the chromosome. Zoom slider: Zooms in and out on a chromosome. 36
37 Change Display Options IGV offers several display op;ons for tracks Zoom in and Zoom out Modify Track Height Sort the Tracks Filter the Tracks Group the Tracks Sort Tracks based on Region of Interest 37
38 Variants Visualization in IGV Load TAIR10 genome to IGV Load BAM file SRR bam to IGV with Load from URL to- BAM_BAM.bam Load VCF file var.raw.vcf to IGV with Load from URL 38
39 Zoom in to : Chr5:57,073-57,142 Zoom In Screen 39
40 Variants Visualization in IGV Load TAIR10 genome to IGV Load BAM file SRR bam to IGV with Load from URL Load VCF file var.raw.vcf to IGV with Load from URL hxp://biocluster.ucr.edu/~rkaundal/galaxy_workshop/snpseq/gatk.vcf 40
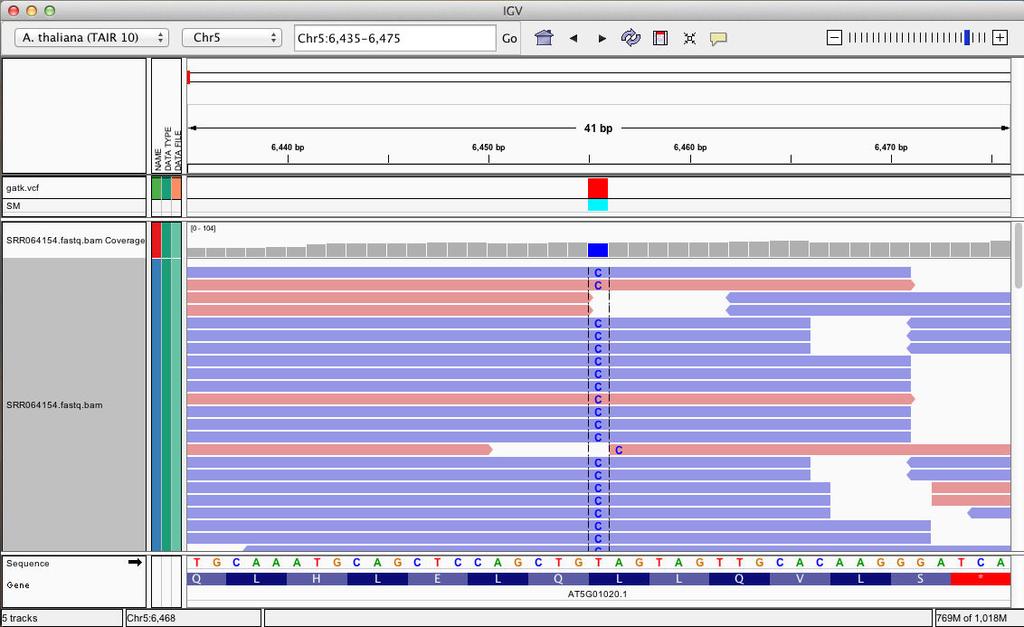
41 Zoom in position (chr5:6,435-6,475)
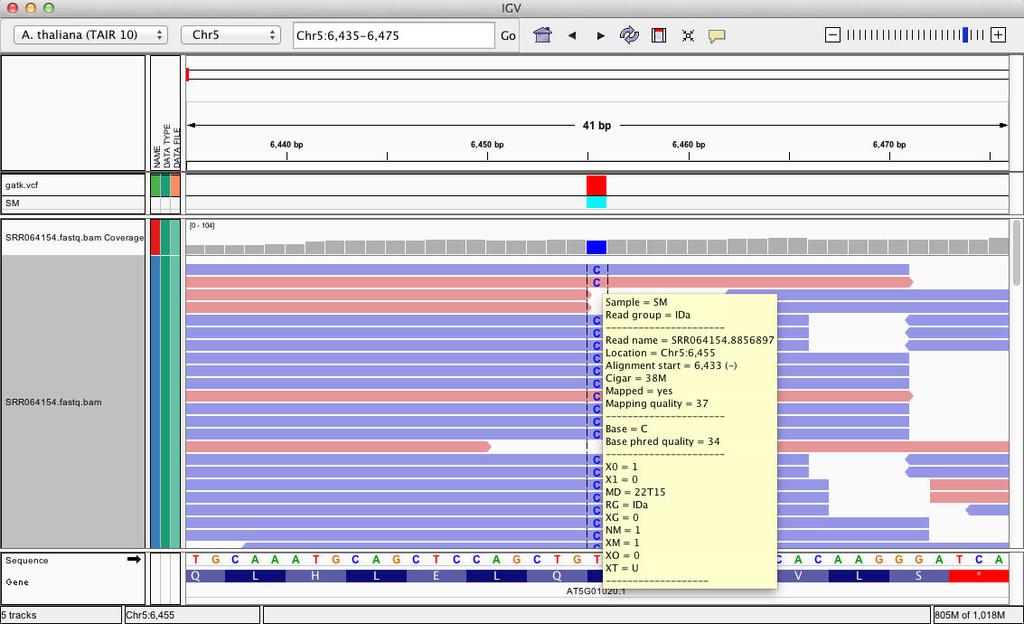
42 Zoom in position (chr5:6,435-6,475)

43 Right click on track for more options
44 Show all bases in IGV
45 RNA-seq Results Visualization Load four BAM files to IGV Load gene differen;al expression GFF3 file expression_diff.gff3 to IGV 44

46 Exercise 2: RNA-seq Result Visualization Zoom in to Chr1:41,351-51,208

47 Exercise 2: RNA-seq Result Visualization Zoom in Chr1:49,457-51,457
48 UCR Galaxy homepage (
49 Thank You
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Maize genome sequence in FASTA format. Gene annotation file in gff format
 Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
Exercise 1. Using Tophat/Cufflinks to analyze RNAseq data. Step 1. One of CBSU BioHPC Lab workstations has been allocated for your workshop exercise. The allocations are listed on the workshop exercise
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Genome 373: Mapping Short Sequence Reads III. Doug Fowler
 Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Read Mapping and Variant Calling
 Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
RNAseq analysis: SNP calling. BTI bioinformatics course, spring 2013
 RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
RNA-Seq Analysis With the Tuxedo Suite
 June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
June 2016 RNA-Seq Analysis With the Tuxedo Suite Dena Leshkowitz Introduction In this exercise we will learn how to analyse RNA-Seq data using the Tuxedo Suite tools: Tophat, Cuffmerge, Cufflinks and Cuffdiff.
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
de.nbi and its Galaxy interface for RNA-Seq
 de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
de.nbi and its Galaxy interface for RNA-Seq Jörg Fallmann Thanks to Björn Grüning (RBC-Freiburg) and Sarah Diehl (MPI-Freiburg) Institute for Bioinformatics University of Leipzig http://www.bioinf.uni-leipzig.de/
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
Read mapping with BWA and BOWTIE
 Read mapping with BWA and BOWTIE Before We Start In order to save a lot of typing, and to allow us some flexibility in designing these courses, we will establish a UNIX shell variable BASE to point to
Read mapping with BWA and BOWTIE Before We Start In order to save a lot of typing, and to allow us some flexibility in designing these courses, we will establish a UNIX shell variable BASE to point to
TP RNA-seq : Differential expression analysis
 TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
Practical exercises Day 2. Variant Calling
 Practical exercises Day 2 Variant Calling Samtools mpileup Variant calling with samtools mpileup + bcftools Variant calling with HaplotypeCaller (GATK Best Practices) Genotype GVCFs Hard Filtering Variant
Practical exercises Day 2 Variant Calling Samtools mpileup Variant calling with samtools mpileup + bcftools Variant calling with HaplotypeCaller (GATK Best Practices) Genotype GVCFs Hard Filtering Variant
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Using Galaxy: RNA-seq
 Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
Using Galaxy: RNA-seq Stanford University September 23, 2014 Jennifer Hillman-Jackson Galaxy Team Penn State University http://galaxyproject.org/ The Agenda Introduction RNA-seq Example - Data Prep: QC
SAMtools. SAM BAM. mapping. BAM sort & indexing (ex: IGV) SNP call
 SNP call") SAMtools http://samtools.sourceforge.net/ SAM/BAM mapping BAM SAM BAM BAM sort & indexing (ex: IGV) mapping SNP call SAMtools NGS Program: samtools (Tools for alignments in the SAM format) Version: 0.1.19
SAMtools http://samtools.sourceforge.net/ SAM/BAM mapping BAM SAM BAM BAM sort & indexing (ex: IGV) mapping SNP call SAMtools NGS Program: samtools (Tools for alignments in the SAM format) Version: 0.1.19
Variant calling using SAMtools
 Variant calling using SAMtools Calling variants - a trivial use of an Interactive Session We are going to conduct the variant calling exercises in an interactive idev session just so you can get a feel
Variant calling using SAMtools Calling variants - a trivial use of an Interactive Session We are going to conduct the variant calling exercises in an interactive idev session just so you can get a feel
Evaluate NimbleGen SeqCap RNA Target Enrichment Data
 Roche Sequencing Technical Note November 2014 How To Evaluate NimbleGen SeqCap RNA Target Enrichment Data 1. OVERVIEW Analysis of NimbleGen SeqCap RNA target enrichment data generated using an Illumina
Roche Sequencing Technical Note November 2014 How To Evaluate NimbleGen SeqCap RNA Target Enrichment Data 1. OVERVIEW Analysis of NimbleGen SeqCap RNA target enrichment data generated using an Illumina
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers
 Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
AgroMarker Finder manual (1.1)
") AgroMarker Finder manual (1.1) 1. Introduction 2. Installation 3. How to run? 4. How to use? 5. Java program for calculating of restriction enzyme sites (TaqαI). 1. Introduction AgroMarker Finder (AMF)is
AgroMarker Finder manual (1.1) 1. Introduction 2. Installation 3. How to run? 4. How to use? 5. Java program for calculating of restriction enzyme sites (TaqαI). 1. Introduction AgroMarker Finder (AMF)is
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Helpful Galaxy screencasts are available at:
 This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Our typical RNA quantification pipeline
 RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
RNA-Seq primer Our typical RNA quantification pipeline Upload your sequence data (fastq) Align to the ribosome (Bow>e) Align remaining reads to genome (TopHat) or transcriptome (RSEM) Make report of quality
NGS : reads quality control
 NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
Analyzing Variant Call results using EuPathDB Galaxy, Part II
 Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
Analyzing Variant Call results using EuPathDB Galaxy, Part II In this exercise, we will work in groups to examine the results from the SNP analysis workflow that we started yesterday. The first step is
replace my_user_id in the commands with your actual user ID
 Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
Services Performed. The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples.
 Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Services Performed The following checklist confirms the steps of the RNA-Seq Service that were performed on your samples. SERVICE Sample Received Sample Quality Evaluated Sample Prepared for Sequencing
Variation among genomes
 Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
From the Schnable Lab:
 From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Identiyfing splice junctions from RNA-Seq data
 Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Identiyfing splice junctions from RNA-Seq data Joseph K. Pickrell pickrell@uchicago.edu October 4, 2010 Contents 1 Motivation 2 2 Identification of potential junction-spanning reads 2 3 Calling splice
Resequencing Analysis. (Pseudomonas aeruginosa MAPO1 ) Sample to Insight
 Sample to Insight") Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
TopHat, Cufflinks, Cuffdiff
 TopHat, Cufflinks, Cuffdiff Andreas Gisel Institute for Biomedical Technologies - CNR, Bari TopHat TopHat TopHat TopHat is a program that aligns RNA-Seq reads to a genome in order to identify exon-exon
TopHat, Cufflinks, Cuffdiff Andreas Gisel Institute for Biomedical Technologies - CNR, Bari TopHat TopHat TopHat TopHat is a program that aligns RNA-Seq reads to a genome in order to identify exon-exon
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6
 Exercise 6") Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
Mapping RNA sequence data (Part 1: using pathogen portal s RNAseq pipeline) Exercise 6 The goal of this exercise is to retrieve an RNA-seq dataset in FASTQ format and run it through an RNA-sequence analysis
Short Read Sequencing Analysis Workshop
 Short Read Sequencing Analysis Workshop Day 1 Introduc.on to the Workshop Schedule for Week 1 Day 1: Introduc.on Workshop syllabus and schedule Basic considera.ons for sequencing depth, read length, format,
Short Read Sequencing Analysis Workshop Day 1 Introduc.on to the Workshop Schedule for Week 1 Day 1: Introduc.on Workshop syllabus and schedule Basic considera.ons for sequencing depth, read length, format,
Handling sam and vcf data, quality control
 Handling sam and vcf data, quality control We continue with the earlier analyses and get some new data: cd ~/session_3 wget http://wasabiapp.org/vbox/data/session_4/file3.tgz tar xzf file3.tgz wget http://wasabiapp.org/vbox/data/session_4/file4.tgz
Handling sam and vcf data, quality control We continue with the earlier analyses and get some new data: cd ~/session_3 wget http://wasabiapp.org/vbox/data/session_4/file3.tgz tar xzf file3.tgz wget http://wasabiapp.org/vbox/data/session_4/file4.tgz
Today's outline. Resources. Genome browser components. Genome browsers: Discovering biology through genomics. Genome browser tutorial materials
 Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Today's outline Genome browsers: Discovering biology through genomics BaRC Hot Topics April 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Genome browser introduction Popular
Next Generation Sequence Alignment on the BRC Cluster. Steve Newhouse 22 July 2010
 Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Decrypting your genome data privately in the cloud
 Decrypting your genome data privately in the cloud Marc Sitges Data Manager@Made of Genes @madeofgenes The Human Genome 3.200 M (x2) Base pairs (bp) ~20.000 genes (~30%) (Exons ~1%) The Human Genome Project
Decrypting your genome data privately in the cloud Marc Sitges Data Manager@Made of Genes @madeofgenes The Human Genome 3.200 M (x2) Base pairs (bp) ~20.000 genes (~30%) (Exons ~1%) The Human Genome Project
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Our data for today is a small subset of Saimaa ringed seal RNA sequencing data (RNA_seq_reads.fasta). Let s first see how many reads are there:
. Let s first see how many reads are there:") Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
Practical Course in Genome Bioinformatics 19.2.2016 (CORRECTED 22.2.2016) Exercises - Day 5 http://ekhidna.biocenter.helsinki.fi/downloads/teaching/spring2016/ Answer the 5 questions (Q1-Q5) according
Part 1: How to use IGV to visualize variants
 Using IGV to identify true somatic variants from the false variants http://www.broadinstitute.org/igv A FAQ, sample files and a user guide are available on IGV website If you use IGV in your publication:
Using IGV to identify true somatic variants from the false variants http://www.broadinstitute.org/igv A FAQ, sample files and a user guide are available on IGV website If you use IGV in your publication:
DNA Sequencing analysis on Artemis
 DNA Sequencing analysis on Artemis Mapping and Variant Calling Tracy Chew Senior Research Bioinformatics Technical Officer Rosemarie Sadsad Informatics Services Lead Hayim Dar Informatics Technical Officer
DNA Sequencing analysis on Artemis Mapping and Variant Calling Tracy Chew Senior Research Bioinformatics Technical Officer Rosemarie Sadsad Informatics Services Lead Hayim Dar Informatics Technical Officer
Handling important NGS data formats in UNIX Prac8cal training course NGS Workshop in Nove Hrady 2014
 Handling important NGS data formats in UNIX Prac8cal training course NGS Workshop in Nove Hrady 2014 Vaclav Janousek, Libor Morkovsky hjp://ngs- course- nhrady.readthedocs.org (Exercises & Reference Manual)
Handling important NGS data formats in UNIX Prac8cal training course NGS Workshop in Nove Hrady 2014 Vaclav Janousek, Libor Morkovsky hjp://ngs- course- nhrady.readthedocs.org (Exercises & Reference Manual)
Introduction to Galaxy
 Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
Introduction to Galaxy Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW Day 1 Thurs 28 th January 2016 Overview What is Galaxy? Description of
Scalable RNA Sequencing on Clusters of Multicore Processors
 JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
JOAQUÍN DOPAZO JOAQUÍN TARRAGA SERGIO BARRACHINA MARÍA ISABEL CASTILLO HÉCTOR MARTÍNEZ ENRIQUE S. QUINTANA ORTÍ IGNACIO MEDINA INTRODUCTION DNA Exon 0 Exon 1 Exon 2 Intron 0 Intron 1 Reads Sequencing RNA
ChIP-Seq Tutorial on Galaxy
 1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
1 Introduction ChIP-Seq Tutorial on Galaxy 2 December 2010 (modified April 6, 2017) Rory Stark The aim of this practical is to give you some experience handling ChIP-Seq data. We will be working with data
Welcome to GenomeView 101!
 Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Welcome to GenomeView 101! 1. Start your computer 2. Download and extract the example data http://www.broadinstitute.org/~tabeel/broade.zip Suggestion: - Linux, Mac: make new folder in your home directory
Introduction to Galaxy
 Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
Introduction to Galaxy Saint Louis University St. Louis, Missouri April 30, 2013 Dave Clements, Emory University http://galaxyproject.org/ Agenda 9:00 Welcome 9:20 Basic Analysis with Galaxy 10:30 Basic
Supplementary Figure 1. Fast read-mapping algorithm of BrowserGenome.
 Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu
 GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
GSNAP: Fast and SNP-tolerant detection of complex variants and splicing in short reads by Thomas D. Wu and Serban Nacu Matt Huska Freie Universität Berlin Computational Methods for High-Throughput Omics
NGS Analyses with Galaxy
 1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
Importing your Exeter NGS data into Galaxy:
 Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Genome representa;on concepts. Week 12, Lecture 24. Coordinate systems. Genomic coordinates brief overview 11/13/14
 2014 - BMMB 852D: Applied Bioinforma;cs Week 12, Lecture 24 István Albert Biochemistry and Molecular Biology and Bioinforma;cs Consul;ng Center Penn State Genome representa;on concepts At the simplest
2014 - BMMB 852D: Applied Bioinforma;cs Week 12, Lecture 24 István Albert Biochemistry and Molecular Biology and Bioinforma;cs Consul;ng Center Penn State Genome representa;on concepts At the simplest
WM2 Bioinformatics. ExomeSeq data analysis part 1. Dietmar Rieder
 WM2 Bioinformatics ExomeSeq data analysis part 1 Dietmar Rieder RAW data Use putty to logon to cluster.i med.ac.at In your home directory make directory to store raw data $ mkdir 00_RAW Copy raw fastq
WM2 Bioinformatics ExomeSeq data analysis part 1 Dietmar Rieder RAW data Use putty to logon to cluster.i med.ac.at In your home directory make directory to store raw data $ mkdir 00_RAW Copy raw fastq
NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab
![NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab](/thumbs/74/70633133.jpg "NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab") NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
Supplementary Information. Detecting and annotating genetic variations using the HugeSeq pipeline
 Supplementary Information Detecting and annotating genetic variations using the HugeSeq pipeline Hugo Y. K. Lam 1,#, Cuiping Pan 1, Michael J. Clark 1, Phil Lacroute 1, Rui Chen 1, Rajini Haraksingh 1,
Supplementary Information Detecting and annotating genetic variations using the HugeSeq pipeline Hugo Y. K. Lam 1,#, Cuiping Pan 1, Michael J. Clark 1, Phil Lacroute 1, Rui Chen 1, Rajini Haraksingh 1,
The software comes with 2 installers: (1) SureCall installer (2) GenAligners (contains BWA, BWA- MEM).
 SureCall installer (2) GenAligners (contains BWA, BWA- MEM).") Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Pre-processing and quality control of sequence data. Barbera van Schaik KEBB - Bioinformatics Laboratory
 Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
m6aviewer Version Documentation
 m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
m6aviewer Version 1.6.0 Documentation Contents 1. About 2. Requirements 3. Launching m6aviewer 4. Running Time Estimates 5. Basic Peak Calling 6. Running Modes 7. Multiple Samples/Sample Replicates 8.
RNA Sequencing with TopHat and Cufflinks
 RNA Sequencing with TopHat and Cufflinks Introduction 3 Run TopHat App 4 TopHat App Output 5 Run Cufflinks 18 Cufflinks App Output 20 RNAseq Methods 27 Technical Assistance ILLUMINA PROPRIETARY 15050962
RNA Sequencing with TopHat and Cufflinks Introduction 3 Run TopHat App 4 TopHat App Output 5 Run Cufflinks 18 Cufflinks App Output 20 RNAseq Methods 27 Technical Assistance ILLUMINA PROPRIETARY 15050962
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Gene expression estimation
 Gene expression estimation") mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
mrna-seq Basic processing Read mapping (shown here, but optional. May due if time allows) Tophat Gene expression estimation cufflinks Confidence intervals Gene expression changes (separate use case) Sample
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
RNASeq2017 Course Salerno, September 27-29, 2017
 RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
RNASeq2017 Course Salerno, September 27-29, 2017 RNA- seq Hands on Exercise Fabrizio Ferrè, University of Bologna Alma Mater (fabrizio.ferre@unibo.it) Hands- on tutorial based on the EBI teaching materials
ChIP-seq practical: peak detection and peak annotation. Mali Salmon-Divon Remco Loos Myrto Kostadima
 ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
Introduction to Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide
 RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
RNA Sequencing with TopHat Alignment v1.0 and Cufflinks Assembly & DE v1.1 App Guide For Research Use Only. Not for use in diagnostic procedures. Introduction 3 Set Analysis Parameters TopHat 4 Analysis
Reference guided RNA-seq data analysis using BioHPC Lab computers
 Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Reference guided RNA-seq data analysis using BioHPC Lab computers This document assumes that you already know some basics of how to use a Linux computer. Some of the command lines in this document are
Copy Number Variations Detection - TD. Using Sequenza under Galaxy
 Copy Number Variations Detection - TD Using Sequenza under Galaxy I. Data loading We will analyze the copy number variations of a human tumor (parotid gland carcinoma), limited to the chr17, from a WES
Copy Number Variations Detection - TD Using Sequenza under Galaxy I. Data loading We will analyze the copy number variations of a human tumor (parotid gland carcinoma), limited to the chr17, from a WES
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Aligners. J Fass 21 June 2017
 Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
Aligners J Fass 21 June 2017 Definitions Assembly: I ve found the shredded remains of an important document; put it back together! UC Davis Genome Center Bioinformatics Core J Fass Aligners 2017-06-21
INTRODUCTION AUX FORMATS DE FICHIERS
 INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Using the Galaxy Local Bioinformatics Cloud at CARC
 Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University
Using the Galaxy Local Bioinformatics Cloud at CARC Lijing Bu Sr. Research Scientist Bioinformatics Specialist Center for Evolutionary and Theoretical Immunology (CETI) Department of Biology, University