Cycle «Analyse de données de séquençage à haut-débit» Module 1/5 Analyse ADN. Sophie Gallina CNRS Evo-Eco-Paléo (EEP)
|
|
|
- Candace Wood
- 6 years ago
- Views:
Transcription




 (sophie.")
1 Cycle «Analyse de données de séquençage à haut-débit» Module 1/5 Analyse ADN Sophie Gallina CNRS Evo-Eco-Paléo (EEP)
2 Module 1/5 Analyse DNA NGS Introduction Galaxy : upload data, datasets & histories Reads Quality Control Reads cleaning Aligning reads on reference Galaxy Workflow & best practices March / 15
3 NGS introduction Sequencers Librairies, adaptors Multiplexing, barcode Reads, single-end, paired-end Encoding quality with scores Fastq Format Cleaning reads Analysis protocoles (alignment and assembly) March / 15
4 Sequencers Illumina Source : March / 15
5 Libraries Source : Thierry Grange, 5ème Ecole de bioinformatique AVIESAN-IFB DNA fragment (size depends on libraries) March / 15
6 Adaptators Source : Thierry Grange, 5ème Ecole de bioinformatique AVIESAN-IFB ements/eba216 Roscoff_216_Grange_.pdf DNA fragment without adaptator DNA fragment with adaptators March / 15
7 Read = DNA fragment end Single-end Read 1 Sequencing only 1 end Paired-end Sequencing both ends Reads orientation Insert size Read 1 DNA fragment DNA fragment Read 2 Insert size Based on DNA fragment size, these situations may arise : DNA fragment Read 1 Read 1 Read 2 March 217 DNA fragment Read 2 7 / 15
8 Multiplexing, barcode ex Illumina HiSeq2 Adaptators within DNA fragment are sequenced. Adaptators are removed, they are not present in files provided by sequencing platforms. When used for multiplexing, sequences are demultiplexed in differents files. DNA fragment without adaptator DNA fragment with adaptators March / 15
9 Sequencer's output : fastq file format 1 read Identifiant Sequence CGCCCGGCCAATCATTGTGGTTTTAAGTCACTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCT + CTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCTCTGTGCACCCAGCATTGCCCAGAACAGGGC + CTCCCAGCTTCCAACAGACCCTGTCCCAGCTCCCTCCAAGCTGAGTGTTGGCCTGATACCTACCAGTGGAGCGAGGGGAACCCGAGGACTGCCAAGGGCA + AAAAAAAAAAAAAAAAAAAAAAAAAAAAAGGGGGCCCCCCTTTCCCCCCCGGGGGGGGGACAGGGGGGGTGTTCGGGCCCCGCGCCGCCCTTGACCACGG + EKLMPPPPPQQQQQQQQQQQQQQQK########################################################################### March / 15
10 Paired-end fastq file format 2 files : Forward (1) Reverse (2) 1 interleaved paired CTAGGAAGCGTAGTCCTGGGGTCATCTCTCCTATTAATACTGTTGGGGAATGTTTAGTA + CATTATTTCATAGTAGCCAAAAAGTGGAAACAGTCAAAATATCCGTCAGTGAATTGACC + 1./.,/&((&3=;B@F86C>@51(3:).6GG TATTTCTGGAATTTTCCATTTAATATTTTCAGACTGCAGTTGACTGCGGGTAACTGAAA + TTCTGGTCAGTAAGACCTCAAAAGGTTAAATACTAGCGATTTACACACCTTAAATGATT + CCTAAAATGGTGTGTTTTCGTATATTCACAATGCTGTGGAACCATCACCACTATCTGAT + TCTTTCTTTTGTTTTTTTTTCTGAGATGTCTTTTGTTTTTGTTCTGAGGTCTTGTTATG + ILLUMINA_13:3:111:1249:1993 length=11 TTTTCAGAGTAGTTGGTACCCAATTGGAAGATGTGACCCACTTCGATACCGCGCTTGAG + ILLUMINA_13:3:111:1249:1993 length=99 ANNNNNNCTTCGGTATNAACTGGGGNNNNGATGTTGAACTGGGTAAAGTCGAAGATCTG + ILLUMINA_13:3:111:1463:1964 length=11 NTGAGTAGCTCAATGCGCTGACGCCAATAGCTATACCAACGACTGGCCAGATTATGTTT + ILLUMINA_13:3:111:1463:1964 length=99 AAGTGACCCATCGCGATAAAGTGCTGCGCAGTAAANAGCANCTGTTNGATGCTGGCTTA + ILLUMINA_13:3:111:1366:197 length=11 NAAGTCGCGGCGACCCCTATCGTGGCTTTCGGCGTACGCCATTTCAATGCGGCCGCCGC + B[[X[YY[YVcc_cccc_cc ILLUMINA_13:3:111:1366:197 length=99 TGGTCAATACAAGCCGCAATACCTGCATCATGCGGNGGAANAATTTGCGCGCCGTTTTC + ggfegggggggdeggggfgcgggagggggggega^bb`^]b[y[[[zffffh_afeefe March / 15
11 Reads quality Errors when reading bases Depends on sequencing technologie Error rate increases with read size For each position in the read One base (ATCG) One error probability March / 15
12 Phred Quality Score (for a base) Phred quality score Q = logarithmically related to the base calling error probabilities P. (Error probabilities depends on sequencing technologie) Source : Score coding : number [-6] => 1 character [more compact in a file] A T C A With coding convention 38=G, 39=H ATCA HHGH There are more than one coding convention for the score (for history reasons) March / 15
13 Quality score coding For history reasons More than one way to compute score More than one coding convention Code ASCII Each keyboard symbole has a number examples!=33, A=65 Source : Galaxy : Always uses Sanger coding => conversion tool (groomer) March / 15
14 Example for score interpretation using sanger coding (Galaxy) Bad : -19 correct : 2-29 Good : 29-4 S - Sanger Phred+33 SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS...!"#$%&'()+,-./ :;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{ }~ March / 15
15 Interpreting quality CGCCCGGCCAATCATTGTGGTTTTAAGTCACTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCT + CTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCTCTGTGCACCCAGCATTGCCCAGAACAGGGC + CTCCCAGCTTCCAACAGACCCTGTCCCAGCTCCCTCCAAGCTGAGTGTTGGCCTGATACCTACCAGTGGAGCGAGGGGAACCCGAGGACTGCCAAGGGCA + AAAAAAAAAAAAAAAAAAAAAAAAAAAAAGGGGGCCCCCCTTTCCCCCCCGGGGGGGGGACAGGGGGGGTGTTCGGGCCCCGCGCCGCCCTTGACCACGG + EKLMPPPPPQQQQQQQQQQQQQQQK########################################################################### SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS...!"#$%&'()+,-./ :;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{ }~ March / 15
16 Read cleaning : CGCCCGGCCAATCATTGTGGTTTTAAGTCACTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCT + CTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCTCTGTGCACCCAGCATTGCCCAGAACAGGGC + CTCCCAGCTTCCAACAGACCCTGTCCCAGCTCCCTCCAAGCTGAGTGTTGGCCTGATACCTACCAGTGGAGCGAGGGGAACCCGAGGACTGCCAAGGGCA + AAAAAAAAAAAAAAAAAAAAAAAAAAAAAGGGGGCCCCCCTTTCCCCCCCGGGGGGGGGACAGGGGGGGTGTTCGGGCCCCGCGCCGCCCTTGACCACGG + CGCCCGGCCAATCATTGTGGTTTTAAGTCACTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCT + CTAAGTTTGAGGCTATTTTGTTTTACAGCAAAAGCTAACTGATGCAGACAGGGACAAGTCAGTCTCATCTCTGTGCACCCAGCATTGCCCAGAACAGGGC + CTCCCAGCTTCCAACAGACCCTGTCCCAGCTCCCTCCAAGCTGAG + D?KMPQEPGCPQQNPQIQIGR@DPERQHEKBED=HCHG8EHFDCD March / 15


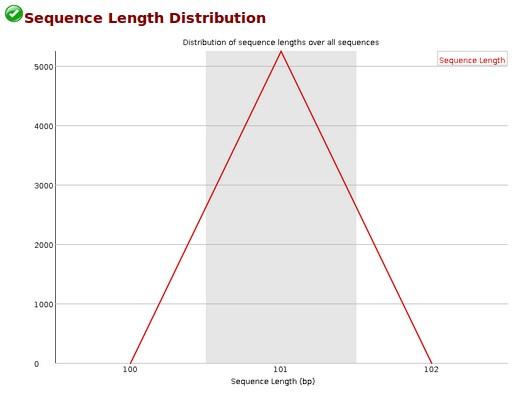
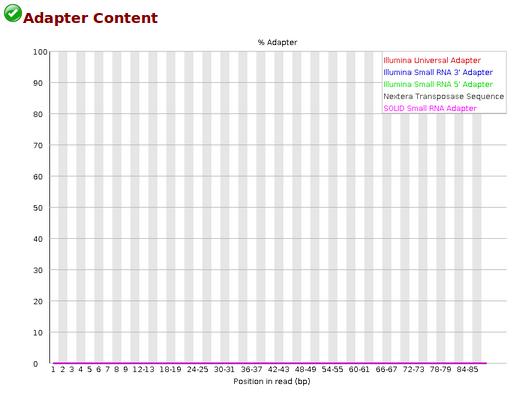
17 Quality control examples March / 15
18 Reads cleaning Cut adaptators at read ends Trimming : cut read ends (5' ou 3') Fixed number of bases Individual base quality Mean quality of bases in a sliding window Filtering : remove read Size criteria (example < 6bp) Mean base quality for all bases criteria (example < 25) March / 15
19 Reads cleaning example : protocole for de-novo transcriptome assembly Clean adaptators Trimming 5' et 3' on base quality (> 3) Trimming using sliding window (4 bases, Q < 2) Filtering on mean read quality (Q < 25) Filtering on read size (taille < 2) Source : Erwan Core, 5ème Ecole de bioinformatique AVIESAN-IFB March / 15
20 Protocole for variant analysis Reads (fastq) Quality control Filtering adaptators, filtering and trimming on base quality Reads cleaning Reads (fastq) Genome (fasta) Quality control Alignement Alignment (sam/bam) Quality control Alignment cleaning Alignment (sam/bam) Filtering on alignment quality, marking duplicated reads, local realignment Metrics, cover statistics Variant detection Variants (vcf) Module 4 cycle NGS Variants cleaning and annotation March / 15
21 Protocole for de-novo assemby Reads (fastq) Quality control Reads cleaning Filtrering adaptators, quality filtering and trimming Reads (fastq) Quality control Assembly Contigs, scaffolds Metrics Assembly cleaning Contigs, scaffolds Metrics March / 15
22 Quality scores Score for base A Score for base C Score for base T Score for base A Read A C T A... Reference Mean Score for read A Alignment score Sample1 Sample2 Score for variant calling : A/T A T A T A T Score for sample genotyping : A/T A T A Score for sample genotyping : NA March / 15
23 Quality scores Quality = probability of no mistake on Base calling Read alignment Variant calling Sample genotyping Depends on algorithm or on protocole Protocole Illumina # protocole PacBio Protocole Illumina v1 # protocole Illumina v2 Instrument's evolution Computation type Probability depends on instrument bias Base calling algorithm # alignment algorithm # variant detection algorithm Differents tools for the same operation BWA # Bowtie2 for alignment March / 15
24 Galaxy Connection Upload data Working with datasets and histories Adding local reference Converting to fastqsanger format March / 15
25 Data for this tutorial Data from Human genome from Hapmap project Reference : small region from chromosome 2 2:38-53 (assembly GRCh37) file GRCh37_region1.fasta Reads: Illumina paired-end (2x1bp) for 3 samples (HG96, HG11 and HG13) files HGXXX_1.fastq, HGXXX_2.fastq (only reads for this small region, for reasons of speed) Dowload files on billile wkiki : Main goals for this first part of tutorial Upload reference and reads for one sample (HG11) Work with histories, datasets and tools March / 15




26 Connect : Galaxy v1 1 Enter IP number 2 Click on Galaxy icon IP simple IP + session 3 Menu User / login 4 Username : user@galaxy.ifb.fr Password : ifbuser Username + Password 5 Menu User => Check that you are connected March / 15



27 Connect : Galaxy v2 1 Enter IP number 2 Menu User / login 3 Username : bilillen Password : bilillen 4 Menu User => Check that you are connected March / 15

28 History : «Folder» containing a set of data Default name = «Unamed history» 1 Rename history => TP1 2 Explore history menu 3 Create new history March / 15

29 List histories, go back to TP1 1 List all histories 2 Go back to TP1 history March / 15
30 Dataset ~ «Data file» Upload reference in a dataset 1 Tools Get Data / Upload File 3 fasta 2 Choose file 4 unspecified 5 start 6 close 2 Choose file GRCh37_regions1.fasta 3 Choose fasta format (! not csfasta) 4 Keep «Unspecified» as genome 5 Run with start 6 Close March / 15




31 Dataset : summary, attributes, full data 1 Click on dataset name show summary of attributes and data 2 Click on the eye show data 3 Click on pencil show attributes March / 15

32 Add a local reference (TP_ref) 1 Menu User / Custom Builds 2 2 Choose name TP_ref 3 Choose fasta format 4 Choose dataset n 1 : GRCh37_regions1.fasta 5 Submit Reference is now available March / 15


33 Check / Change database attribute 1 Analyse Data 2 1 Menu Analyze Data 2 Click on dataset name to see summary => database attribute is «?» 3 Click on pencil to change attributes 4 Choose TP_ref database 5 Save 6 check database attribute is now «TP_ref» March / 15

34 Upload reads (fastq) for sample HG11 1 Tools: Get Data / Upload File 2 Choose files HG11_1_fastq and HG11_2.fastq 3 Choose «fastq» format 4 choose «TP_ref» genome 5 Run with start 6 Close 7 Check attributes fastq 2 choose local file 4 TP_ref 5 start 6 close March / 15

35 Look at a fastq file identifiant sequence quality 1 read What is the file size? How many reads? Sizes of reads? Which is the quality coding? Galaxy : always uses Sanger => conversion tool : groomer Source : March / 15


36 Convert to Sanger format : groomer tool Tools: FASTQ Groomer 2 Choose to «groom» many files 3 Choose files HG11_1_fastq and HG11_2.fastq 4 Choose «Sanger & Illumina 1.8+» format 5 Execute Create 2 new datasets (N 4 et 5) 6 Check new datasets attributes What are the sizes of new datasets? How many reads? Which is the quality coding? What are the names of new datasets? March / 15


37 Rename datasets 2 HG11_OK_1.fastq For each new datasets : 1 Click on pencil to change attributes 2 Change the name 3 Save 4 Check new datasets names After changin a dataset name, how can we retreive dataset origin? March / 15

38 Retreive dataset origin This dataset results from groomer tool, applied on dataset 2 (HG11_1.fastq) March / 15
39 Reads quality control Per base quality Per read mean quality Read size Adaptators Duplicated reads March / 15


40 Reads quality control (fastqc) Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data ,2 Choose tool : FastQC 3 Choose datasets n 4 et 5 4 Execute Create 4 new datasets For each fastq file : 1 «raw data» and 1 «Webpage» 3 4 March / 15


 2 Remove")


41 Manage fastqc result datasets Look quickly at dataset content (we will deeply look at that later) 2 Remove «RawData» datasets 3 Rename «Webpage»datasets HG11_1.QC et HG11_2.QC March / 15


42 FastqQC : Summary & Basic Statistics March / 15

43 FastQC : Per base sequence quality Median Mean Good Quality Mean Quality Bad quality March / 15
44 Fasqc : Per base sequence quality Example OK Example KO Source : bad_sequence_fastqc.html March / 15

45 Fastqc : Per sequence quality score Example OK Example KO Source : bad_sequence_fastqc.html March / 15


46 FastQC : Sequence Length Distribution & Per sequence GC content March / 15


47 FastQC : Per base sequence content Example OK Example KO Source : bad_sequence_fastqc.html March / 15


48 FastQC : Per base N content Example OK Example KO Source : bad_sequence_fastqc.html March / 15

49 FastQC : Overrepresented sequences Example OK Example KO Source : bad_sequence_fastqc.html March / 15


50 FastQC : Sequence Duplication Levels Example OK Example KO Source : bad_sequence_fastqc.html March / 15


51 FastQC Adapter Content Example OK Example KO Source : bad_sequence_fastqc.html March / 15

52 FastQC Kmer Content Example OK Example KO Source : bad_sequence_fastqc.html March / 15


53 FastQC Example with PacBio Source : pacbio_srr7514_fastqc.html March / 15
54 Cleaning Reads Filtrering adaptators Filtering & trimming reads Comparing quality before and after cleaning March / 15
55 Filtering & trimming Filtering = remove reads Based on quality or size criteria Trimming = remove read ends Fixed number of bases Bases < quality March / 15


56 Trimming Cut bad quality bases at the end of reads Exemple OK Exemple KO Source : bad_sequence_fastqc.html March / 15

57 Filtering Remove reads with bad mean quality Exemple OK Exemple KO Source : bad_sequence_fastqc.html March / 15
, pp.")
58 Reads cleaning (Trimmomatic) 1/2 Bolger, A. M. and Lohse, M. and Usadel, B. (214). Trimmomatic: a flexible trimmer for Illumina sequence data. In Bioinformatics, 3 (15), pp Choose files 2 Parameters for adaptators March / 15

59 Reads cleaning (Trimmomatic) 2/2 Add operations (cleaning steps) : 1 : LEADING : Cut bad quality 5' bases 2 : TRAILNG:Cut bad quality 3' bases 3 : SLIDINGWINDOW : Cut bases with bad mean quality in a sliding window 4 : AVGQUAL : remove reads with bad mean quality 5 : MINLEN : Remove small size reads March / 15
 Reads2 after cleaning Reads1 after cleaning How many")

60 Trimmomatic : Results Unpaired reads2 (corresponding reads1 has been removed during cleaning) Unpaired reads1 (corresponding reads2 has been removed during cleaning) Reads2 after cleaning Reads1 after cleaning How many paired reads after cleaning? Are there any trace or log of what happens during cleaning? From summary, click on «i» icon Look content of «stdout» This contains all messages send by this tool during execution. March / 15

61 Trimmomatic : 2nd try Run this tool again after changing AVGQUAL parameter value to Use one of the datasets produced by previous analysis 2 Click on «Run this job again» icon 2 All parameters are pre-sets with values used in the previous execution 3 3 Change only the parameter in step n 4 AVGQUAL value 25 How many paired reads after new cleaning? Work only with paired dataset produced by first cleaning (remove second try) Rename the datatsets HG11_1_clean.fastq and HG11_2_clean.fastq Run quality control on these 2 datasets. Compare quality control before and after cleaning March / 15


62 Compare quality control before / after cleaning Solution 1 : Open a second web browser window Connect to access history and datasets Visualize datasets HG11_OK_1.CQ HG11_clean_1.QC March / 15

63 Compare quality control before / after cleaning Solution 2 : Use Galaxy «Scratchbook» to manage Galaxy windows 1 «Enable Scratchbook» 2 Visualize dataset HG11_clean_1.fastq 3 Visualize dataset HG11_OK_1.fastq March / 15



64 Quality control before & after cleaning HG11_OK_1.QC HG11_clean_1.QC March / 15



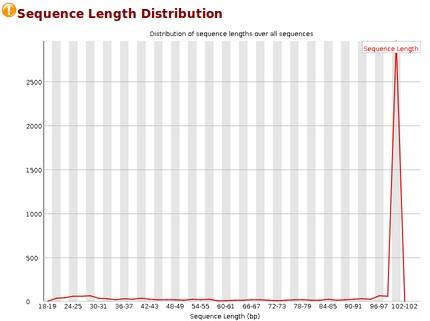
65 Quality control before & after cleaning HG11_OK_1.QC HG11_clean_1.QC March / 15
66 Reads alignment SAM/BAM Format SAM results for single-end and paired-end reads BWA & Bowtie2 alignment tools Manage duplicated reads (picard / MarkDuplicates) Count alignments (samtools / flagstat) Compute deepth and coverage (Deeptools/PlotCoverage) March / 15
67 SAM - Sequence Alignment/Map Alignement Coding to a SAM format SN:ref LN:45 r1 99 ref 7 r2 ref 9 r3 ref 9 r4 ref 16 r3 264 ref 29 r1 147 ref M2I4M1D3M 3S6M1P1I4M 5S6M 6M14N5M 6H5M 9M = = 7 39 TTAGATAAAGGATACTG AAAAGATAAGGATA GCCTAAGCTAA ATAGCTTCAGC TAGGC CAGCGGCAT SA:Z:ref,29,,6H5M,17,; SA:Z:ref,9,+,5S6M,3,1; NM:i:1 Source : March / 15

68 SAM File format : Reference sequence SN:ref : sequence name, LN:45 : sequence size 1 line per read, 12 SN:ref LN:45 r1 99 ref 7 r2 ref 9 r3 ref 9 r4 ref 16 r3 264 ref 29 r1 147 ref M2I4M1D3M 3S6M1P1I4M 5S6M 6M14N5M 6H5M 9M = = 7 39 TTAGATAAAGGATACTG AAAAGATAAGGATA GCCTAAGCTAA ATAGCTTCAGC TAGGC CAGCGGCAT SA:Z:ref,29,,6H5M,17,; SA:Z:ref,9,+,5S6M,3,1; NM:i:1 1 log 1 ( p ) P = error probability on position Mapping tools estimation based on missmatch, insertions, deletions multiple alignements Source : March / 15
69 SAM - SN:ref LN:45 r1 99 ref 7 r2 ref 9 r3 ref 9 r4 ref 16 r3 264 ref 29 r1 147 ref 37 1 : + 2 : + 32 : + 64 : M2I4M1D3M 3S6M1P1I4M 5S6M 6M14N5M 6H5M 9M = = 7 39 TTAGATAAAGGATACTG AAAAGATAAGGATA GCCTAAGCTAA ATAGCTTCAGC TAGGC CAGCGGCAT SA:Z:ref,29,,6H5M,17,; SA:Z:ref,9,+,5S6M,3,1; NM:i:1 template having multiple segments in sequencing each segment properly aligned according to the aligner SEQ of the next segment in the template being reverse complemented the first segment in the template Source : March / 15

70 SAM - SN:ref LN:45 r1 99 ref 7 r2 ref 9 r3 ref 9 r4 ref 16 r3 264 ref 29 r1 147 ref M2I4M1D3M 3S6M1P1I4M 5S6M 6M14N5M 6H5M 9M = = 7 39 TTAGATAAAGGATACTG AAAAGATAAGGATA GCCTAAGCTAA ATAGCTTCAGC TAGGC CAGCGGCAT SA:Z:ref,29,,6H5M,17,; SA:Z:ref,9,+,5S6M,3,1; NM:i:1 March / 15
71 BAM : Binary sam Same data as in SAM «binary» format, more compact Smaller files Faster treatment for computers BAI : Index for BAM file Speed up data search and retrieve in a BAM file March / 15
72 SAM Examples for single-end reads S1 S2 S3 S4 S5 S6 S7 S8 S9 S1 S11 S12 S13 S14 S15 S16 S lecture parfaite, bonne qualité lecture parfaite, mauvaise qualité lecture parfaite, bonne qualité sauf sur les 2 dernières bases lecture parfaite, bonne qualité sauf sur les 6 dernières bases insertion de 5 bases en position 5, bonne qualité deletion de 5 bases en position 5, bonne qualité 5 substitutions réparties sur le read, bonne qualité 5 substitutions au début du read, bonne qualité 5 substitutions au milieu du read, bonne qualité 5 substitutions au début, mauvaise qualité lecture parfaite, bonne qualité, duplicaion 1 lecture parfaite, bonne qualité, duplication 2 lecture parfaite, bonne qualité lecture parfaite, bonne qualité, décalé de 1bp lecture non alignée, bonne qualité lecture alignée 2 fois, bonne qualité lecture alignée 1 fois, bonne qualité SAM flags read paired read mapped in proper pair read unmapped mate unmapped read reverse strand mate reverse strand first in pair second in pair not primary alignment read fails platform/vendor quality checks read is PCR or optical duplicate supplementary alignment BWA QNAME S1 S2 S3 S4 S5 S6 S7 S8 S9 S1 S12 S12 S13 S14 S15 S16 S17 FLAG 4 RNAME POS MAPQ x x Bowtie2 CIGAR 1M 1M 1M 1M 48M5I47M 5M5D45M 1M 5S95M 1M 5S95M 1M 1M 1M 99M 1M 1M NEXT PNEXT TLEN QNAME S1 S2 S3 S4 S5 S6 S7 S8 S9 S1 S12 S12 S13 S14 S15 S16 S17 March 217 FLAG 4 RNAME POS MAPQ CIGAR 1M 1M 1M 1M 48M5I47M 5M5D45M 1M 4M1I95M 1M 4M1I95M 1M 1M 1M 99M 1M 1M NEXT PNEXT TLEN 72 / 15
73 SAM Examples for single-end reads S1 S2 S3 S4 S5 S6 S7 S8 S9 S1 S11 S12 S13 S14 S15 S16 S lecture parfaite, bonne qualité lecture parfaite, mauvaise qualité lecture parfaite, bonne qualité sauf sur les 2 dernières bases lecture parfaite, bonne qualité sauf sur les 6 dernières bases insertion de 5 bases en position 5, bonne qualité deletion de 5 bases en position 5, bonne qualité 5 substitutions réparties sur le read, bonne qualité 5 substitutions au début du read, bonne qualité 5 substitutions au milieu du read, bonne qualité 5 substitutions au début, mauvaise qualité lecture parfaite, bonne qualité, duplicaion 1 lecture parfaite, bonne qualité, duplication 2 lecture parfaite, bonne qualité lecture parfaite, bonne qualité, décalé de 1bp lecture non alignée, bonne qualité lecture alignée 2 fois, bonne qualité lecture alignée 1 fois, bonne qualité SAM flags read paired read mapped in proper pair read unmapped mate unmapped read reverse strand mate reverse strand first in pair second in pair not primary alignment read fails platform/vendor quality checks read is PCR or optical duplicate supplementary alignment BWA QNAME FLAG RNAME S1 S2 S3 Duplicated reads S4 S5 S6 S7 S8 S9 S1 S12 S12 S13 S14 S15 4 S16 S17 POS MAPQ x x Bowtie2 CIGAR 1M 1M 1M 1M 48M5I47M 5M5D45M 1M 5S95M 1M 5S95M 1M 1M 1M 99M 1M 1M NEXT PNEXT TLEN Score 6 / Unaligned reads QNAME FLAG S1 S2 Score [-42] S3 S4 S5 S6 S7 S8 S9 S1 S12 S12 S13 S14 S15 4 S16 S17 March 217 RNAME POS MAPQ CIGAR 1M 1M 1M 1M 48M5I47M 5M5D45M 1M 4M1I95M 1M 4M1I95M 1M 1M 1M 99M 1M 1M NEXT PNEXT TLEN Multiple Alignements 73 / 15
74 SAM : Examples for paired-end reads BWA QNAME FLAG RNAME POS MAPQ CIGAR P1_F 97 C M P1_R 145 C M P2_F 97 C M P2_R 145 C M P3_F 73 C M P3_R 133 C1 51 P4_F 97 C M P4_R 145 C M P5_F 97 C M P5_R 145 C M P6_F 97 C M P6_R 145 C M P7_F 65 C M P7_R 129 C M Bowtie2 RNEXT PNEXT TLEN = 41 3 = 21-3 = = 31-9 = 51 = 51 C2 11 C1 61 = = = = 81-2 = = C1 QNAME FLAG RNAME POS MAPQ CIGAR P1_F 99 C M P1_R 147 C M P2_F 97 C M P2_R 145 C M P3_F 73 C M P3_R 133 C1 51 P4_F 97 C M P4_R 145 C M P5_F 99 C M P5_R 147 C M P6_F 99 C M P6_R 147 C M P7_F 65 C M R7_R 129 C M 1 C2 R1 R2 R3 R4 R5 R6 R RNEXT PNEXT TLEN = 41 3 = 21-3 = = 31-9 = 51 = 51 C2 11 C1 61 = 91 3 = 71-3 = 11 3 = 81-3 = = SAM flags read paired x x x x x x x x read mapped in proper pair x x read unmapped x mate unmapped x read reverse strand mate reverse strand x x x x first in pair x x x x second in pair x x x x not primary alignment read fails platform/vendor quality checks read is PCR or optical duplicate supplementary alignment March / 15
75 SAM : Examples for paired-end reads Difference on flags BWA QNAME FLAG RNAME POS MAPQ CIGAR P1_F 97 C M P1_R 145 C M P2_F 97 C M P2_R 145 C M P3_F 73 C M P3_R 133 C1 51 P4_F 97 C M P4_R 145 C M P5_F 97 C M P5_R 145 C M P6_F 97 C M P6_R 145 C M P7_F 65 C M P7_R 129 C M RNEXT PNEXT TLEN = 41 3 = 21-3 = = 31-9 = 51 = 51 C2 11 C1 61 = = = = 81-2 = = Bowtie2 QNAME FLAG RNAME POS MAPQ CIGAR P1_F 99 C M P1_R 147 C M P2_F 97 C M P2_R 145 C M P3_F 73 C M P3_R 133 C1 51 P4_F 97 C M P4_R 145 C M P5_F 99 C M P5_R 147 C M P6_F 99 C M P6_R 147 C M P7_F 65 C M R7_R 129 C M Difference on positions when duplicated sequences C1 R1 R2 R3 R4 R5 R6 R7 1 C2 F R Bowtie2 R RNEXT PNEXT TLEN = 41 3 = 21-3 = = 31-9 = 51 = 51 C2 11 C1 61 = 91 3 = 71-3 = 11 3 = 81-3 = = SAM flags read paired x x x x x x x x read mapped in proper pair x x read unmapped x mate unmapped x read reverse strand mate reverse strand x x x x first in pair x x x x second in pair x x x x not primary alignment read fails platform/vendor quality checks read is PCR or optical duplicate supplementary alignment BWA March / 15


76 Alignment with BWA Li, H. and Durbin, R. (29). Fast and accurate short read alignment with Burrows-Wheeler transform. In Bioinformatics, 25 (14), pp Li, H. and Durbin, R. (21). Fast and accurate longread alignment with Burrows-Wheeler transform. In Bioinformatics, 26 (5), pp March / 15
, pp. R25. Langmead, Ben and Salzberg, Steven L (212).")
77 Alignment with bowtie2 Langmead, Ben and Trapnell, Cole and Pop, Mihai and Salzberg, Steven L (29). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. In Genome Biology, 1 (3), pp. R25. Langmead, Ben and Salzberg, Steven L (212). Fast gapped-read alignment with Bowtie 2. In Nature Methods, 9 (4), pp March / 15

78 Cleaning duplicated reads Source : GATK Marking duplicates March / 15




79 Picard / MarkDuplicate Additional information about Picard tools is available from Picard web site at BWA Bowtie2 UNPAIRED_READS_EXAMINED READ_PAIRS_EXAMINED SECONDARY_OR_SUPPLEMENTARY_RDS 3 UNMAPPED_READS UNPAIRED_READ_DUPLICATES READ_PAIR_DUPLICATES READ_PAIR_OPTICAL_DUPLICATES PERCENT_DUPLICATION,2962,296 ESTIMATED_LIBRARY_SIZE March / 15
 8126 + paired in sequencing 463 + read1 463 + read2 798 + properly paired (98.2%: nan%) 888 + with itself and mate mapped 19 + singletons (.")
 8126 + paired in sequencing 463 + read1 463 + read2 874 + properly paired (99.36%: nan%) 886 + with itself and mate mapped 18 + singletons (.")
80 Alignment count : samtools flagstat BWA in total (QC passed reads + QC failed reads) + secondary 3 + supplementary 24 + duplicates mapped (99.77%: nan%) paired in sequencing read read properly paired (98.2%: nan%) with itself and mate mapped 19 + singletons (.23%: nan%) + with mate mapped to a different chr + with mate mapped to a different chr (mapq>=5) Bowtie in total (QC passed reads + QC failed reads) + secondary + supplementary 24 + duplicates mapped (99.73%: nan%) paired in sequencing read read properly paired (99.36%: nan%) with itself and mate mapped 18 + singletons (.22%: nan%) + with mate mapped to a different chr + with mate mapped to a different chr (mapq>=5) March / 15
81 Coverage and deepth of coverage Source : Élodie Girard, 5ème Ecole de bioinformatique AVIESAN-IFB March / 15


82 Computing coverage and deepth of coverage DeepTools2 / plotcoverage Ramírez, Fidel and Ryan, Devon P and Grüning, Björn and Bhardwaj, Vivek and Kilpert, Fabian and Richter, Andreas S and Heyne, Steffen and Dündar, Friederike and Manke, Thomas (216). deeptools2: a next generation web server for deep-sequencing data analysis. In Nucleic Acids Research, 44 (W1), pp. W16 W165 March / 15


83 DeepTools / Plot Coverage March / 15
84 Galaxy Workflow Extract workflow from an history Modify workflow Execute workflow on new data Compare results from 2 workflows (in 2 histories) March / 15


85 Extract Workflow from the history of steps applied to the first sample March / 15

86 Visualize workflow March / 15

87 Modify workflow visualisation March / 15


88 Modify some steps configuration This WF uses 3 input files. Change box name to describe which data is required for each input : eg Reference, Forward fastq, Reverse fastq You can also change any parameter for example for trimmomatic step. March / 15


89 Enable a parameter to be set at run time Parameters for each tool will have the predefined values set in the workflow You can modify this to enable any parameter to be set at run time. Modify Trimmomatic so that Adapter are set at run time Do'nt forget to save your workflow! March / 15

90 Import new data for sample HG13 Importe files HG13_1.fastq and HG_13_2.fastq March / 15


91 Analyze these new data with the same workflow Run the workflow with these new data March / 15


92 Browse results March / 15


93 Gather BWA alignment results for the 2 samples 1/2 Create a new history named results From history TP1 : Copy HG11_BWA_MD.bam dataset March / 15



94 Gather BWA alignment results for the 2 samples 2/2 From history HG13 : Copy dataset «MarkDuplicates on data 13: MarkDuplicates BAM output»... March / 15

95 Visualize deepth of coverage for both samples Rename datasets Run plotcoverage March / 15
96 Galaxy Best Practices Manage disk space Export analysis results (datasets and histories) Export / Import analysis protocoles (workflow) March / 15


97 Manage disk space Global disk space Disk space per dataset Disk space per history March / 15




98 Export analysis results : datasets Image Text Bam March 217 HTML + files 98 / 15

99 Export analysis results : histories March / 15

100 Export / import analysis protocoles : workflow Export Import March / 15
101 Galaxy - Visualisation Create a visualisation Add datasets Change visualisation parameters March / 15

102 Create a new visualisation 1 Menu Visualisation / New Track Browser 2 Choose name TP2_vizu 3 Choose reference TP2_ref 4 Create Save March / 15

103 Visualize BAM files 1/3 1 Add datasets 2 Choose BAM files 3 Add March / 15

104 Visualize BAM files 2/3 Zoom and move March / 15


105 Visualize BAM files 3/3 1 Set Display Mode 2 Coverage March / 15
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
INTRODUCTION AUX FORMATS DE FICHIERS
 INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
The SAM Format Specification (v1.3 draft)
") The SAM Format Specification (v1.3 draft) The SAM Format Specification Working Group July 15, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
The SAM Format Specification (v1.3 draft) The SAM Format Specification Working Group July 15, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
The SAM Format Specification (v1.3-r837)
") The SAM Format Specification (v1.3-r837) The SAM Format Specification Working Group November 18, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited
The SAM Format Specification (v1.3-r837) The SAM Format Specification Working Group November 18, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited
NGS : reads quality control
 NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
SAM : Sequence Alignment/Map format. A TAB-delimited text format storing the alignment information. A header section is optional.
 Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Pre-processing and quality control of sequence data. Barbera van Schaik KEBB - Bioinformatics Laboratory
 Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Mar%n Norling. Uppsala, November 15th 2016
 Mar%n Norling Uppsala, November 15th 2016 What can we do with an assembly? Since we can never know the actual sequence, or its varia%ons, valida%ng an assembly is tricky. But once you ve used all the assemblers,
Mar%n Norling Uppsala, November 15th 2016 What can we do with an assembly? Since we can never know the actual sequence, or its varia%ons, valida%ng an assembly is tricky. But once you ve used all the assemblers,
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
NGS Analyses with Galaxy
 1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
1 NGS Analyses with Galaxy Introduction Every living organism on our planet possesses a genome that is composed of one or several DNA (deoxyribonucleotide acid) molecules determining the way the organism
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
File Formats: SAM, BAM, and CRAM. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
TP RNA-seq : Differential expression analysis
 TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
TP RNA-seq : Differential expression analysis Overview of RNA-seq analysis Fusion transcripts detection Differential expresssion Gene level RNA-seq Transcript level Transcripts and isoforms detection 2
Analyzing ChIP- Seq Data in Galaxy
 Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
Analyzing ChIP- Seq Data in Galaxy Lauren Mills RISS ABSTRACT Step- by- step guide to basic ChIP- Seq analysis using the Galaxy platform. Table of Contents Introduction... 3 Links to helpful information...
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
From fastq to vcf. NGG 2016 / Evolutionary Genomics Ari Löytynoja /
 From fastq to vcf Overview of resequencing analysis samples fastq fastq fastq fastq mapping bam bam bam bam variant calling samples 18917 C A 0/0 0/0 0/0 0/0 18969 G T 0/0 0/0 0/0 0/0 19022 G T 0/1 1/1
From fastq to vcf Overview of resequencing analysis samples fastq fastq fastq fastq mapping bam bam bam bam variant calling samples 18917 C A 0/0 0/0 0/0 0/0 18969 G T 0/0 0/0 0/0 0/0 19022 G T 0/1 1/1
Genome 373: Mapping Short Sequence Reads III. Doug Fowler
 Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
Sequence Alignment/Map Format Specification
 Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 2 Sep 2016 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 2 Sep 2016 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
Next Generation Sequence Alignment on the BRC Cluster. Steve Newhouse 22 July 2010
 Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
The SAM Format Specification (v1.4-r956)
") The SAM Format Specification (v1.4-r956) The SAM Format Specification Working Group April 12, 2011 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
The SAM Format Specification (v1.4-r956) The SAM Format Specification Working Group April 12, 2011 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab
![NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab](/thumbs/74/70633133.jpg "NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab") NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1
 EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
RCAC. Job files Example: Running seqyclean (a module)
") RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
Sequence Mapping and Assembly
 Practical Introduction Sequence Mapping and Assembly December 8, 2014 Mary Kate Wing University of Michigan Center for Statistical Genetics Goals of This Session Learn basics of sequence data file formats
Practical Introduction Sequence Mapping and Assembly December 8, 2014 Mary Kate Wing University of Michigan Center for Statistical Genetics Goals of This Session Learn basics of sequence data file formats
SAMtools. SAM BAM. mapping. BAM sort & indexing (ex: IGV) SNP call
 SNP call") SAMtools http://samtools.sourceforge.net/ SAM/BAM mapping BAM SAM BAM BAM sort & indexing (ex: IGV) mapping SNP call SAMtools NGS Program: samtools (Tools for alignments in the SAM format) Version: 0.1.19
SAMtools http://samtools.sourceforge.net/ SAM/BAM mapping BAM SAM BAM BAM sort & indexing (ex: IGV) mapping SNP call SAMtools NGS Program: samtools (Tools for alignments in the SAM format) Version: 0.1.19
The SAM Format Specification (v1.4-r994)
") The SAM Format Specification (v1.4-r994) The SAM Format Specification Working Group January 27, 2012 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
The SAM Format Specification (v1.4-r994) The SAM Format Specification Working Group January 27, 2012 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
Sequence Alignment/Map Format Specification
 Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 28 Feb 2014 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 28 Feb 2014 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
Run Setup and Bioinformatic Analysis. Accel-NGS 2S MID Indexing Kits
 Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
Understanding and Pre-processing Raw Illumina Data
 Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Helpful Galaxy screencasts are available at:
 This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data.
 These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data. We have a few different choices for running jobs on DT2 we will explore both here. We need to alter
These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data. We have a few different choices for running jobs on DT2 we will explore both here. We need to alter
SAM / BAM Tutorial. EMBL Heidelberg. Course Materials. Tobias Rausch September 2012
 SAM / BAM Tutorial EMBL Heidelberg Course Materials Tobias Rausch September 2012 Contents 1 SAM / BAM 3 1.1 Introduction................................... 3 1.2 Tasks.......................................
SAM / BAM Tutorial EMBL Heidelberg Course Materials Tobias Rausch September 2012 Contents 1 SAM / BAM 3 1.1 Introduction................................... 3 1.2 Tasks.......................................
Exeter Sequencing Service
 Exeter Sequencing Service A guide to your denovo RNA-seq results An overview Once your results are ready, you will receive an email with a password-protected link to them. Click the link to access your
Exeter Sequencing Service A guide to your denovo RNA-seq results An overview Once your results are ready, you will receive an email with a password-protected link to them. Click the link to access your
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data
 Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification data Table of Contents Protocol: peak-calling for ChIP-seq data / segmentation analysis for histone modification
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
From the Schnable Lab:
 From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
From the Schnable Lab: Yang Zhang and Daniel Ngu s Pipeline for Processing RNA-seq Data (As of November 17, 2016) yzhang91@unl.edu dngu2@huskers.unl.edu Pre-processing the reads: The alignment software
DNA / RNA sequencing
 Outline Ways to generate large amounts of sequence Understanding the contents of large sequence files Fasta format Fastq format Sequence quality metrics Summarizing sequence data quality/quantity Using
Outline Ways to generate large amounts of sequence Understanding the contents of large sequence files Fasta format Fastq format Sequence quality metrics Summarizing sequence data quality/quantity Using
Using Galaxy for NGS Analyses Luce Skrabanek
 Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
Using Galaxy for NGS Analyses Luce Skrabanek Registering for a Galaxy account Before we begin, first create an account on the main public Galaxy portal. Go to: https://main.g2.bx.psu.edu/ Under the User
PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR
 PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR GOAL OF THIS SESSION Assuming that The audiences know how to perform GWAS
PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR GOAL OF THIS SESSION Assuming that The audiences know how to perform GWAS
Sequence Alignment/Map Format Specification
 Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 14 Jul 2017 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
Sequence Alignment/Map Format Specification The SAM/BAM Format Specification Working Group 14 Jul 2017 The master version of this document can be found at https://github.com/samtools/hts-specs. This printing
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14)
") BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
BGGN-213: FOUNDATIONS OF BIOINFORMATICS (Lecture 14) Genome Informatics (Part 1) https://bioboot.github.io/bggn213_f17/lectures/#14 Dr. Barry Grant Nov 2017 Overview: The purpose of this lab session is
Importing your Exeter NGS data into Galaxy:
 Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
Read Mapping. Slides by Carl Kingsford
 Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Contact: Raymond Hovey Genomics Center - SFS
 Bioinformatics Lunch Seminar (Summer 2014) Every other Friday at noon. 20-30 minutes plus discussion Informal, ask questions anytime, start discussions Content will be based on feedback Targeted at broad
Bioinformatics Lunch Seminar (Summer 2014) Every other Friday at noon. 20-30 minutes plus discussion Informal, ask questions anytime, start discussions Content will be based on feedback Targeted at broad
Introduction to Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Read Alignment UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers
 Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Quality assessment of NGS data
 Quality assessment of NGS data Ines de Santiago July 27, 2015 Contents 1 Introduction 1 2 Checking read quality with FASTQC 1 3 Preprocessing with FASTX-Toolkit 2 3.1 Preprocessing with FASTX-Toolkit:
Quality assessment of NGS data Ines de Santiago July 27, 2015 Contents 1 Introduction 1 2 Checking read quality with FASTQC 1 3 Preprocessing with FASTX-Toolkit 2 3.1 Preprocessing with FASTX-Toolkit:
Next generation sequencing: assembly by mapping reads. Laurent Falquet, Vital-IT Helsinki, June 3, 2010
 Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Read Mapping and Variant Calling
 Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
Bioinformatica e analisi dei genomi
 Bioinformatica e analisi dei genomi Anno 2016/2017 Pierpaolo Maisano Delser mail: maisanop@tcd.ie Background Laurea Triennale: Scienze Biologiche, Universita degli Studi di Ferrara, Dr. Silvia Fuselli;
Bioinformatica e analisi dei genomi Anno 2016/2017 Pierpaolo Maisano Delser mail: maisanop@tcd.ie Background Laurea Triennale: Scienze Biologiche, Universita degli Studi di Ferrara, Dr. Silvia Fuselli;
Next Generation Sequencing quality trimming (NGSQTRIM)
") Next Generation Sequencing quality trimming (NGSQTRIM) Danamma B.J 1, Naveen kumar 2, V.G Shanmuga priya 3 1 M.Tech, Bioinformatics, KLEMSSCET, Belagavi 2 Proprietor, GenEclat Technologies, Bengaluru 3
Next Generation Sequencing quality trimming (NGSQTRIM) Danamma B.J 1, Naveen kumar 2, V.G Shanmuga priya 3 1 M.Tech, Bioinformatics, KLEMSSCET, Belagavi 2 Proprietor, GenEclat Technologies, Bengaluru 3
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection
 CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
ChIP-seq (NGS) Data Formats
 Data Formats") ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
ChIP-seq (NGS) Data Formats Biological samples Sequence reads SRA/SRF, FASTQ Quality control SAM/BAM/Pileup?? Mapping Assembly... DE Analysis Variant Detection Peak Calling...? Counts, RPKM VCF BED/narrowPeak/
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
REPORT. NA12878 Platinum Genome. GENALICE MAP Analysis Report. Bas Tolhuis, PhD GENALICE B.V.
 REPORT NA12878 Platinum Genome GENALICE MAP Analysis Report Bas Tolhuis, PhD GENALICE B.V. INDEX EXECUTIVE SUMMARY...4 1. MATERIALS & METHODS...5 1.1 SEQUENCE DATA...5 1.2 WORKFLOWS......5 1.3 ACCURACY
REPORT NA12878 Platinum Genome GENALICE MAP Analysis Report Bas Tolhuis, PhD GENALICE B.V. INDEX EXECUTIVE SUMMARY...4 1. MATERIALS & METHODS...5 1.1 SEQUENCE DATA...5 1.2 WORKFLOWS......5 1.3 ACCURACY
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Mapping reads to a reference genome
 Introduction Mapping reads to a reference genome Dr. Robert Kofler October 17, 2014 Dr. Robert Kofler Mapping reads to a reference genome October 17, 2014 1 / 52 Introduction RESOURCES the lecture: http://drrobertkofler.wikispaces.com/ngsandeelecture
Introduction Mapping reads to a reference genome Dr. Robert Kofler October 17, 2014 Dr. Robert Kofler Mapping reads to a reference genome October 17, 2014 1 / 52 Introduction RESOURCES the lecture: http://drrobertkofler.wikispaces.com/ngsandeelecture
Falcon Accelerated Genomics Data Analysis Solutions. User Guide
 Falcon Accelerated Genomics Data Analysis Solutions User Guide Falcon Computing Solutions, Inc. Version 1.0 3/30/2018 Table of Contents Introduction... 3 System Requirements and Installation... 4 Software
Falcon Accelerated Genomics Data Analysis Solutions User Guide Falcon Computing Solutions, Inc. Version 1.0 3/30/2018 Table of Contents Introduction... 3 System Requirements and Installation... 4 Software
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Tutorial: De Novo Assembly of Paired Data
 : De Novo Assembly of Paired Data September 20, 2013 CLC bio Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com support@clcbio.com : De Novo Assembly
: De Novo Assembly of Paired Data September 20, 2013 CLC bio Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com support@clcbio.com : De Novo Assembly
Mapping. Reference. read
 Mapping Reference read Assembly vs mapping contig1 contig2 reads bly as s em ll v sa all ma pp all ing vs r efe ren ce Reference What s the problem? Reads differ from the genome due to evolution and sequencing
Mapping Reference read Assembly vs mapping contig1 contig2 reads bly as s em ll v sa all ma pp all ing vs r efe ren ce Reference What s the problem? Reads differ from the genome due to evolution and sequencing
Goal: Learn how to use various tool to extract information from RNAseq reads.
 ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ESSENTIALS OF NEXT GENERATION SEQUENCING WORKSHOP 2017 Class 4 RNAseq Goal: Learn how to use various tool to extract information from RNAseq reads. Input(s): Output(s): magnaporthe_oryzae_70-15_8_supercontigs.fasta
ASAP - Allele-specific alignment pipeline
 ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
Decrypting your genome data privately in the cloud
 Decrypting your genome data privately in the cloud Marc Sitges Data Manager@Made of Genes @madeofgenes The Human Genome 3.200 M (x2) Base pairs (bp) ~20.000 genes (~30%) (Exons ~1%) The Human Genome Project
Decrypting your genome data privately in the cloud Marc Sitges Data Manager@Made of Genes @madeofgenes The Human Genome 3.200 M (x2) Base pairs (bp) ~20.000 genes (~30%) (Exons ~1%) The Human Genome Project
NA12878 Platinum Genome GENALICE MAP Analysis Report
 NA12878 Platinum Genome GENALICE MAP Analysis Report Bas Tolhuis, PhD Jan-Jaap Wesselink, PhD GENALICE B.V. INDEX EXECUTIVE SUMMARY...4 1. MATERIALS & METHODS...5 1.1 SEQUENCE DATA...5 1.2 WORKFLOWS......5
NA12878 Platinum Genome GENALICE MAP Analysis Report Bas Tolhuis, PhD Jan-Jaap Wesselink, PhD GENALICE B.V. INDEX EXECUTIVE SUMMARY...4 1. MATERIALS & METHODS...5 1.1 SEQUENCE DATA...5 1.2 WORKFLOWS......5
CORE Year 1 Whole Genome Sequencing Final Data Format Requirements
 CORE Year 1 Whole Genome Sequencing Final Data Format Requirements To all incumbent contractors of CORE year 1 WGS contracts, the following acts as the agreed to sample parameters issued by NHLBI for data
CORE Year 1 Whole Genome Sequencing Final Data Format Requirements To all incumbent contractors of CORE year 1 WGS contracts, the following acts as the agreed to sample parameters issued by NHLBI for data
NGS FASTQ file format
 NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
NGS FASTQ file format Line1: Begins with @ and followed by a sequence idenefier and opeonal descripeon Line2: Raw sequence leiers Line3: + Line4: Encodes the quality values for the sequence in Line2 (see
Analysis of ChIP-seq data
 Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
Before we start: 1. Log into tak (step 0 on the exercises) 2. Go to your lab space and create a folder for the class (see separate hand out) 3. Connect to your lab space through the wihtdata network and
merantk Version 1.1.1a
 DIVISION OF BIOINFORMATICS - INNSBRUCK MEDICAL UNIVERSITY merantk Version 1.1.1a User manual Dietmar Rieder 1/12/2016 Page 1 Contents 1. Introduction... 3 1.1. Purpose of this document... 3 1.2. System
DIVISION OF BIOINFORMATICS - INNSBRUCK MEDICAL UNIVERSITY merantk Version 1.1.1a User manual Dietmar Rieder 1/12/2016 Page 1 Contents 1. Introduction... 3 1.1. Purpose of this document... 3 1.2. System
Assembly of the Ariolimax dolicophallus genome with Discovar de novo. Chris Eisenhart, Robert Calef, Natasha Dudek, Gepoliano Chaves
 Assembly of the Ariolimax dolicophallus genome with Discovar de novo Chris Eisenhart, Robert Calef, Natasha Dudek, Gepoliano Chaves Overview -Introduction -Pair correction and filling -Assembly theory
Assembly of the Ariolimax dolicophallus genome with Discovar de novo Chris Eisenhart, Robert Calef, Natasha Dudek, Gepoliano Chaves Overview -Introduction -Pair correction and filling -Assembly theory
Resequencing Analysis. (Pseudomonas aeruginosa MAPO1 ) Sample to Insight
 Sample to Insight") Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
Resequencing Analysis (Pseudomonas aeruginosa MAPO1 ) 1 Workflow Import NGS raw data Trim reads Import Reference Sequence Reference Mapping QC on reads Variant detection Case Study Pseudomonas aeruginosa
David Crossman, Ph.D. UAB Heflin Center for Genomic Science. GCC2012 Wednesday, July 25, 2012
 David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
David Crossman, Ph.D. UAB Heflin Center for Genomic Science GCC2012 Wednesday, July 25, 2012 Galaxy Splash Page Colors Random Galaxy icons/colors Queued Running Completed Download/Save Failed Icons Display
Data Preprocessing. Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Practical Bioinformatics for Life Scientists. Week 4, Lecture 8. István Albert Bioinformatics Consulting Center Penn State
 Practical Bioinformatics for Life Scientists Week 4, Lecture 8 István Albert Bioinformatics Consulting Center Penn State Reminder Before any serious work re-check the documentation for small but essential
Practical Bioinformatics for Life Scientists Week 4, Lecture 8 István Albert Bioinformatics Consulting Center Penn State Reminder Before any serious work re-check the documentation for small but essential
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv
 Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
Mapping and Viewing Deep Sequencing Data bowtie2, samtools, igv Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology tan@ciwemb.edu 27 August 2013
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Tutorial. De Novo Assembly of Paired Data. Sample to Insight. November 21, 2017
 De Novo Assembly of Paired Data November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com AdvancedGenomicsSupport@qiagen.com
De Novo Assembly of Paired Data November 21, 2017 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com AdvancedGenomicsSupport@qiagen.com
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
The software comes with 2 installers: (1) SureCall installer (2) GenAligners (contains BWA, BWA- MEM).
 SureCall installer (2) GenAligners (contains BWA, BWA- MEM).") Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional
Release Notes Agilent SureCall 4.0 Product Number G4980AA SureCall Client 6-month named license supports installation of one client and server (to host the SureCall database) on one machine. For additional