Finding homologous sequences in databases
|
|
|
- Hilary Henderson
- 5 years ago
- Views:
Transcription
1 Finding homologous sequences in databases
2 There are multiple algorithms to search sequences databases BLAST (EMBL, NCBI, DDBJ, local) FASTA (EMBL, local) For protein only databases scan via Smith-Waterman alignments 2
3 FASTA Pearson and Lipman 1988 PNAS 85:
4 Aim Look for homologs or similar sequences to a query sequence in a database The search can be performed on: remote computers on the EMBL (or DDBJ) web sites 4
5 Problem: Smith-Waterman is slow. You have a 200bp sequence and you wish to find homologues in the NCBI/Genbank database. But the latter is hundreds of billions - of nucleotides.... to make matters worse, you are searching not for an eact match but rather for something that is related (potentially distantly related) to your query sequence. NCBI receives (as of 2006) over requests for such searches every day. 5
6 Problem: Smith-Waterman is slow. Consider a related problem. To assemble reads into contigs you must align them pairwise to find small overlaps. If you have N = 107 reads (for a genome with 500bp reads, this is enough for a 5 coverage), then you need N(N-1)/2 pairwise comparisons. That is alignments each requiring computations. 6
7 So why not search for larger segments than just one nucleotide A lack of keywords would indicate that there is no need to search further. e.g. For computational biology you might wish to first search for the keywords... nucleotides alignment databases and then subsequently refine the search. If you did such a search in Jane Austen's Sense & Sensibility then you (or the computer) would know not to search any further within this book. 7
8 So why not search for larger segments than just one nucleotide A lack of keywords indicates there is no need to search further. e.g. But you might wish to search for... nucleotides alignment databases If you did such a search in Jane Austen's Sense & Sensibility then you (or the computer) would have to search within this book. Looking for these short segments might etend your search and slow it down 8
9 The K-tuple or Ktup or word size value determine how many consecutive identities are required for a match to be declared For protein searches Ktup = 2 For nucleotide sequences Ktup = 4 or 6 For short sequences (<20nt) Ktup = 1 This value will determine the speed and the sensitivity of the search HASH table: divide the database into a series of tables that contain an ordered (alphabetized) list of words along with links to their location in the database. 9
10 10
11 11
12 12
13 Establishment of a 'table' of sequences of variable length init1 assigned to each regions of a sequence using BLOSUM50 matri to score mismatches 'Table' sorted in alphabetical order initn sum of the init1 of each region Comparison of sequences to query sequence = init1-20 = init1-20 +init if (initn < any init1); the initn is discarded and regions are not joined; initn init1ma opt Score for the Smith -Waterman alignments 13
14 Sequence 1. Identification of regions of identity Query 14
15 Sequence 2. Selection of the region with the best score Query 15
16 3. Join the segments Sequence t1 i in t1 i in t1 i in Query 16
17 4. Compute the new score for the whole sequence initn = init1-20 = init init Sequence t1 i in t1 i in t1 i in Query 17
18 This is a visual (aka vague) description... How does a computer do it? 18
19 A k-tuple listing for sequences J: CCATCGCCATCG I: GCATCGGC K=2 AA AA AC AC AG AG AT AT CA CA CC CC CG CG CT CT GA GA GC GC GG GG GT GT TA TA TC TC TG TG TT TT 19
20 A k-tuple listing for sequences J: CCATCGCCATCG I: GCATCGGC K=2 AA AA AC AC AG AG AT 3 9 AT 3 CA 2 8 CA 2 CC 1 7 CC CG 5 11 CG CT CT GA GA GC GC 1 GG GG 6 GT GT TA TA TC TC TG TG TT TT
21 A k-tuple listing for sequences J: CCATCGCCATCG I: GCATCGGC AA AA AC AC AG AG AT 3 9 AT 3 CA 2 8 CA 2 CC 1 7 CC CG 5 11 CG CT CT GA GA GC 5 GC 1 GG GG 6 GT GT TA TA TC TC TG TG TT TT For GC (i=1) in sequence I, we know there is a match in sequence J at position 6 (j=6). This is on the 1-6=-5 diagonal
22 A k-tuple listing for sequences J: CCATCGCCATCG I: GCATCGGC AA AA AC AC AG AG AT 3 9 AT 3 CA 2 8 CA 2 CC 1 7 CC CG 5 11 CG CT CT GA GA GC 5 GC 1 GG GG 6 GT GT TA TA TC TC TG TG TT TT For GC (i=1) in sequence I, we know there is a match in sequence J at position 6 (j=6). This is on the 1-6=-5 diagonal. 7 For CA (i=2) in sequence I, we know there is a match in sequence J at position 2 (j=2). This is on the 2-2=0 diagonal (the 'main' diagonal). With a single pass through this we can calculate the number of matches on each diagonal. 4 22
23 A k-tuple listing for sequences J: CCATCGCCATCG I: GCATCGGC AA AA AC AC AG AG AT 3 9 AT 3 CA 2 8 CA 2 S0 = 4 (four k-tuples on the 'main' diagonal) CC 1 7 CC CG 5 11 CG 5 S1 = 1 (one k-tuple above the 'main' diagonal) GC 1 S-5 = 1 (one k-tuple 5 below the 'main' diagonal) GG GG 6 GT GT TA TA CT CT GA GA GC TC TC TG TG TT TT 7 S-6 = 4 (four k-tuples 6 below the 'main' diagonal) 4 All this can be done with a single pass through this lookup table. 23
24 Steps for FASTA 1 Calculate k-tuples for query sequence 2 Score diagonals and identify 10 best diagonals 3 Rescore with a scoring matri (allowing for less than k-tuple matches) and identify high scoring regions (init1) 4 - Join the initial regions with joining penalties (initn) 5 - Perform full alignment for sequences with high initn scores (opt). 24
25 How do we know that the match we obtained (the opt score) is unusual? For pairwise alignments we obtained the variation (distribution) to compare our score with from a random permutation. We can't do permutations for every score from every sequence in the databases! So what do we compare just the ''best score to? 25
26 How do we know that the match we obtained (the opt score) is unusual? Originally this was done empirically. Compare just the ''best score to a random collection of alignments (database matches) in order to determine how variable the opt scores are with or without true matches. 26
27 opt score ( )( )( )( )( )( )( )( ) done in discrete units ln(seq. length) Fit a linear regression line. Calculate a z-score. 27
28 opt score ( )( )( )( )( )( )( )( ) done in discrete units ln(seq. length) Z = (score mean)/stddev mean from the linear regression line stddev from the spread (variation) of the points Z - a measure of the no. of stddev above/below the mean 28
29 Obviously this lacks the beauty and simplicity of permutations. If the query or database sequence has unusual amino acid content this is ignored. Other than on a gross scale, differences due to length are ignored. Placing a probability on the Z-score is dubious at best since their distribution is unknown. More recently this is done in a more advanced fashion that is used for BLAST searches. Since this was an advance which came with BLAST we will defer the discussion of this till then. 29
30 Finding protein, nucleic sequences or domains in different databases fasta3: the current main version, it compares DNA query vs. DNA database (K-tuple = 6) or AA query versus AA database (K-tuple = 2) fast3/fasty3: translation of DNA query into the 3 frames AA vs AA database fast3: indels between codons fasty3: indels anywhere 30
31 tfast3/tfasty3: AA query vs DNA translated into 6 frames AA databases fastf3/tfastf3: ordered AA miture (peptide degradation) query to AA or DNA database respectively fasts3/tfasts3: set of fragment AA query (mass spec analysis) to AA or DNA database respectively 31
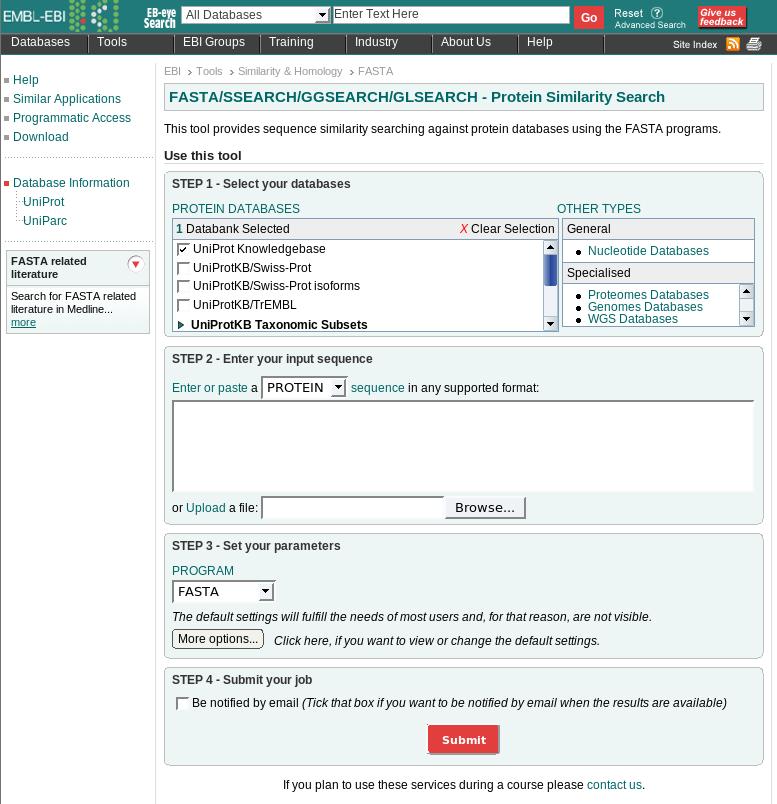
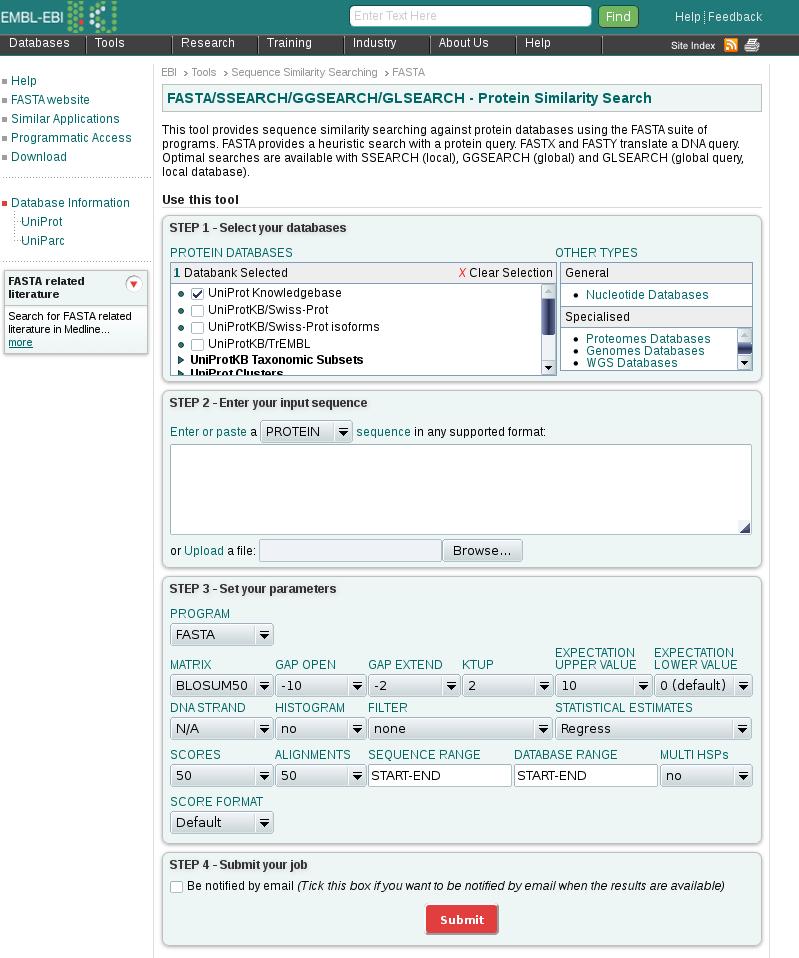
32 Remote query ferredoin gene of Halobacterium halobium Eample : In a to fasta@ebi.ac.uk LIB UNIPROT WORD 1 LIST 50 TITLE HALHA SEQ PTVEYLNYETLDDQGWDMDDDDLFEKAADAGLDGEDYGTMEVAE GEYILEAAEAQGYDWPFSCRAGACANCASIVKEGEIDMDMQQILSD EEVEEKDVRLTCIGSPAADEVKIVYNAKHLDYLQNRVI 32
33 Options first line: data libraries to be search second line: word size or k-tuple value List n: display the top n scores Title: subject of the mail message Align n: align the top n to query sequence ONE: compare only the given strand to the data base Prot: force the query sequence to be a protein PATH: string mails the results back to the string 33
34 Protein search UniProt UniRef100 UniRef90 UniRef50 UniParc swissprot ipi prints sgt pdb imgthlap Euro Patents Japan Patents USPTO Patents A non-redundant collection of all proteins As for UniProt but eliminate identical proteins As for UniProt but eliminate proteins > 90% identical As for UniProt but eliminate proteins > 50% identical As for UniProt but included archived proteins (show changes to an entry) Proteins in the SwissProt database Proteins in the International Protein Inde Proteins in the FingerPrints database Proteins in the Structural Genomics Targets database Proteins in the 3D structural database PDB at Rutgers Proteins in the Immunogenetics Database Proteins in the European patents database Proteins in the Japanese patents database Proteins in the American patents database 34
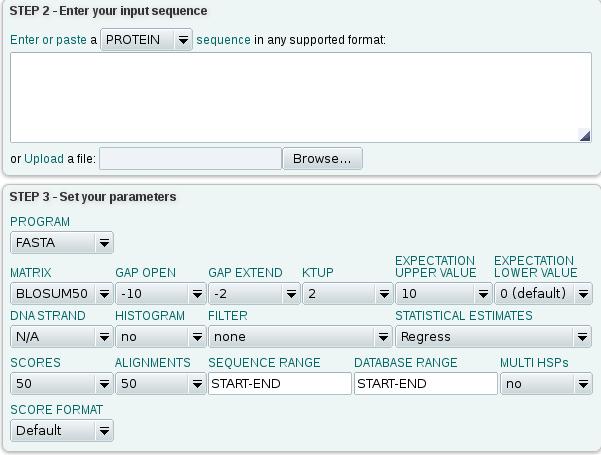
35 Nucleotide search EMBL Fungi INVERTEBRATES HUMAN MAMMALS ORGANELLES BACTERIOPHAGE PLANT PROKARYOTES RODENTS MOUSE STSs SYNTHETIC UNCLASSIFIED VIRUSES VERTEBRATES The entire EMBL database Subsection of EMBL Sequence Tagged Sites 35
36 ESTs GSSs HTGs PATENTS VECTORS EMBLNEW EMBLALL IMGTLIGM IMGTHLA HGVBASE Epressed Sequence Tags Genome Survey Sequences High Throughput Genomics Sequences Sequences new since the last major database release EMBL + EMBLNEW Immunoglobulins and T cells receptors data base Human Major Histocompatibility Comple (MHC/HLA) database Human Genome Variation database 36
37 Web query Direct query on the EMBL websites (or mirrors) Depending on the moment of the day queries could be really slow 37
38 38
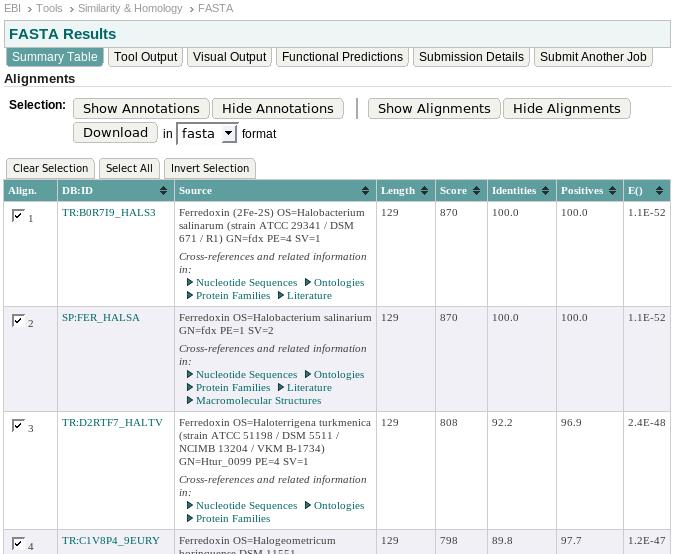
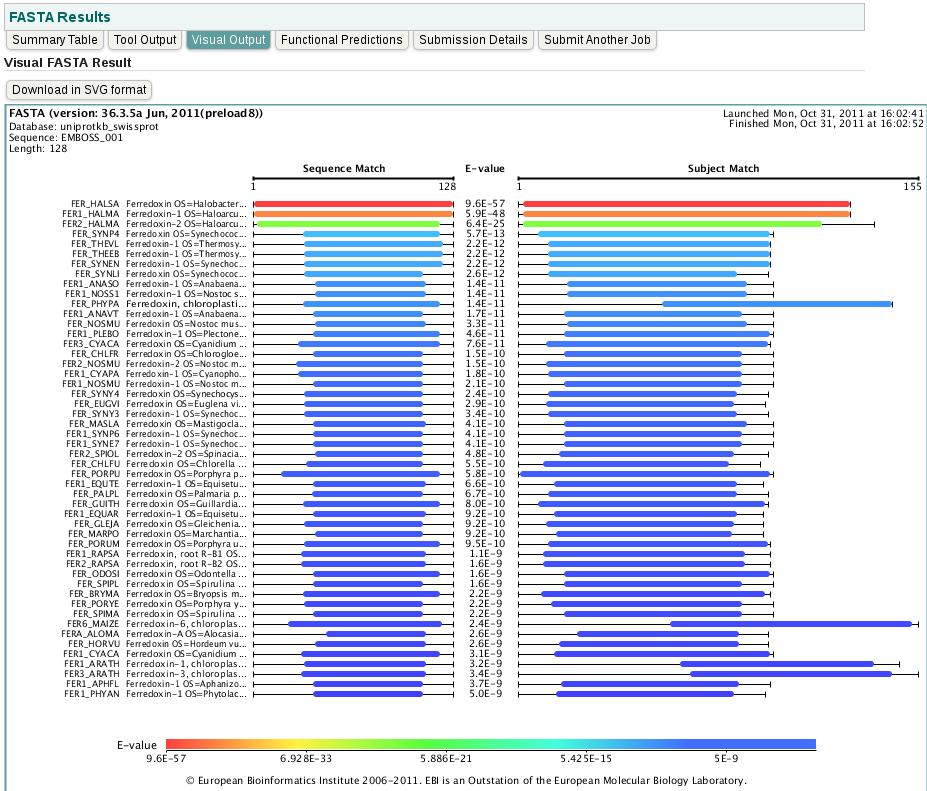
39 39
40 40
41 41
42 Output The output will begin with some information FASTA searches a protein or DNA sequence data bank Version Feb. 20, 2010 Please cite: W.R. Pearson & D.J. Lipman PNAS (1988) 85: >>>EMBOSS_ aa Library: UniProtKB residues in sequences Version References Search libraries sequence type query length 42
43 43
44 44
45 45
46 Output Tabular output Graphical view Listing Histogram (optional): show the number of sequences with various scores 46
47 47
48 48
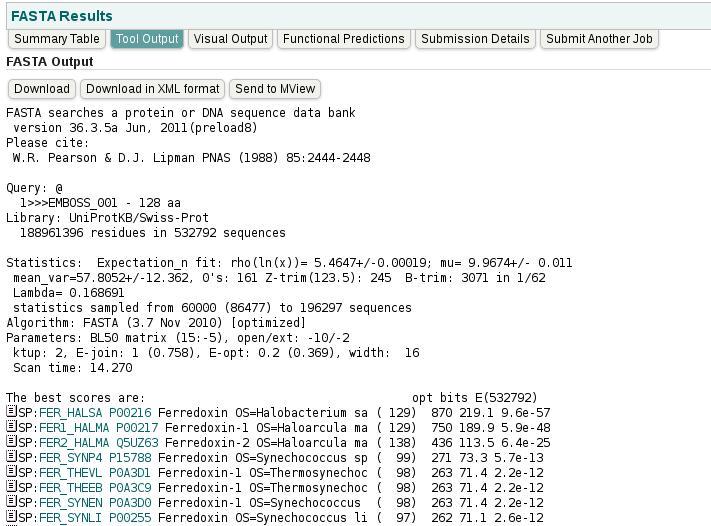
49 49
50 50
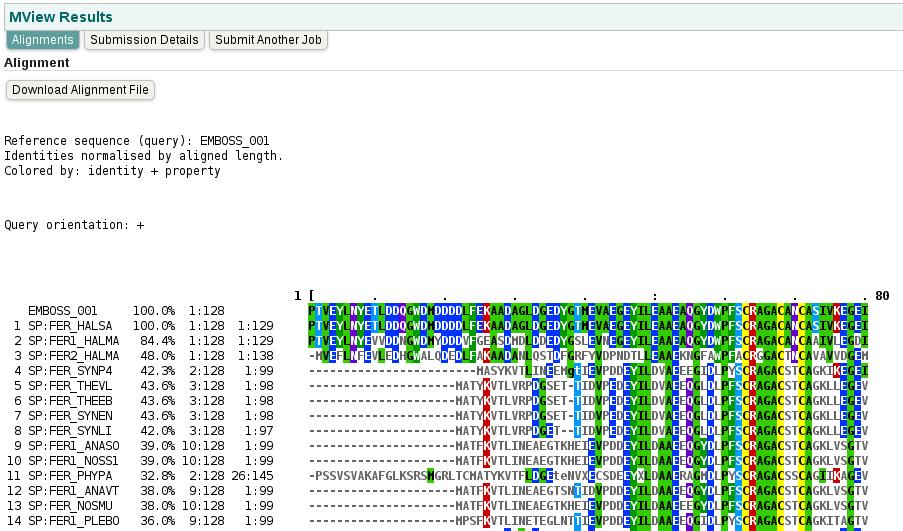
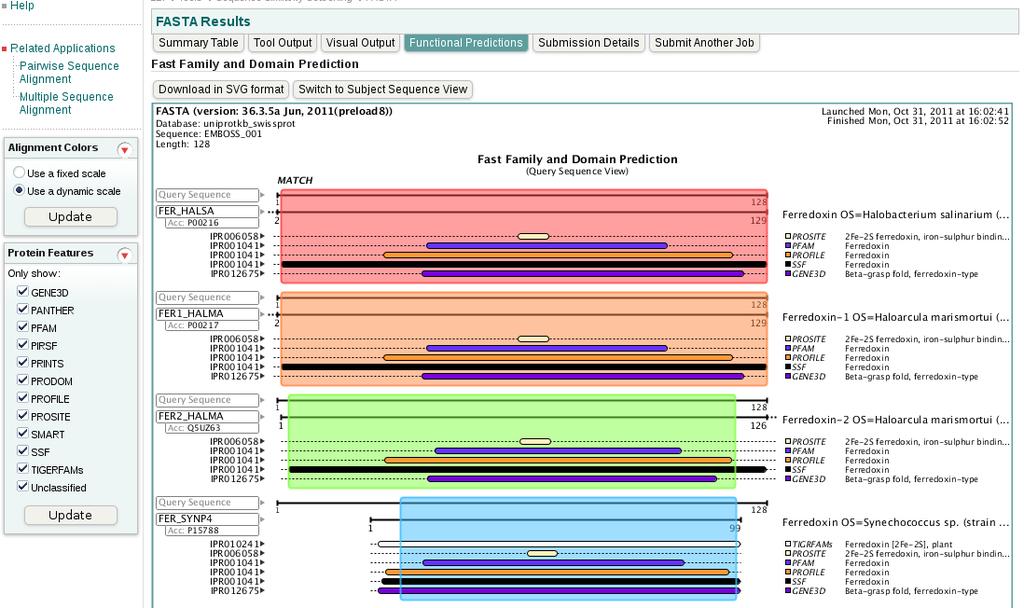
51 51
52 52
53 53
54 54
55 residues in sequences statistics sampled from to sequences Epectation_n fit: rho(ln())= / ; mu= / mean_var= / , 0's: 22 Z-trim: 43 B-trim: 0 in 0/68 Lambda=
56 Score Epected number 56
57 Sequences related to ferredoin 57
58 58
59 59
60 60
61 61
62 62

63 Scores length of the sequence Name of the sequence Reference Number 63
64 64
65 65
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Utility of Sliding Window FASTA in Predicting Cross- Reactivity with Allergenic Proteins. Bob Cressman Pioneer Crop Genetics
 Utility of Sliding Window FASTA in Predicting Cross- Reactivity with Allergenic Proteins Bob Cressman Pioneer Crop Genetics The issue FAO/WHO 2001 Step 2: prepare a complete set of 80-amino acid length
Utility of Sliding Window FASTA in Predicting Cross- Reactivity with Allergenic Proteins Bob Cressman Pioneer Crop Genetics The issue FAO/WHO 2001 Step 2: prepare a complete set of 80-amino acid length
FastA & the chaining problem
 FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:
 FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
Lecture 5 Advanced BLAST
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
EBI patent related services
 EBI patent related services 4 th Annual Forum for SMEs October 18-19 th 2010 Jennifer McDowall Senior Scientist, EMBL-EBI EBI is an Outstation of the European Molecular Biology Laboratory. Overview Patent
EBI patent related services 4 th Annual Forum for SMEs October 18-19 th 2010 Jennifer McDowall Senior Scientist, EMBL-EBI EBI is an Outstation of the European Molecular Biology Laboratory. Overview Patent
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018)
") Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Scoring and heuristic methods for sequence alignment CG 17
 Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
VL Algorithmen und Datenstrukturen für Bioinformatik ( ) WS15/2016 Woche 9
 WS15/2016 Woche 9") VL Algorithmen und Datenstrukturen für Bioinformatik (19400001) WS15/2016 Woche 9 Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Contains material from
VL Algorithmen und Datenstrukturen für Bioinformatik (19400001) WS15/2016 Woche 9 Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Contains material from
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
ICB Fall G4120: Introduction to Computational Biology. Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology
 ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
ICB Fall 2008 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2008 Oliver Jovanovic, All Rights Reserved. The Digital Language of Computers
Chapter 4: Blast. Chaochun Wei Fall 2014
 Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
BLAST. NCBI BLAST Basic Local Alignment Search Tool
 BLAST NCBI BLAST Basic Local Alignment Search Tool http://www.ncbi.nlm.nih.gov/blast/ Global versus local alignments Global alignments: Attempt to align every residue in every sequence, Most useful when
BLAST NCBI BLAST Basic Local Alignment Search Tool http://www.ncbi.nlm.nih.gov/blast/ Global versus local alignments Global alignments: Attempt to align every residue in every sequence, Most useful when
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
EBI services. Jennifer McDowall EMBL-EBI
 EBI services Jennifer McDowall EMBL-EBI The SLING project is funded by the European Commission within Research Infrastructures of the FP7 Capacities Specific Programme, grant agreement number 226073 (Integrating
EBI services Jennifer McDowall EMBL-EBI The SLING project is funded by the European Commission within Research Infrastructures of the FP7 Capacities Specific Programme, grant agreement number 226073 (Integrating
Lecture 4: January 1, Biological Databases and Retrieval Systems
 Algorithms for Molecular Biology Fall Semester, 1998 Lecture 4: January 1, 1999 Lecturer: Irit Orr Scribe: Irit Gat and Tal Kohen 4.1 Biological Databases and Retrieval Systems In recent years, biological
Algorithms for Molecular Biology Fall Semester, 1998 Lecture 4: January 1, 1999 Lecturer: Irit Orr Scribe: Irit Gat and Tal Kohen 4.1 Biological Databases and Retrieval Systems In recent years, biological
Sequence Alignment Heuristics
 Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Biostrings. Martin Morgan Bioconductor / Fred Hutchinson Cancer Research Center Seattle, WA, USA June 2009
 Biostrings Martin Morgan Bioconductor / Fred Hutchinson Cancer Research Center Seattle, WA, USA 15-19 June 2009 Biostrings Representation DNA, RNA, amino acid, and general biological strings Manipulation
Biostrings Martin Morgan Bioconductor / Fred Hutchinson Cancer Research Center Seattle, WA, USA 15-19 June 2009 Biostrings Representation DNA, RNA, amino acid, and general biological strings Manipulation
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame
 1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
1 When we search a nucleic acid databases, there is no need for you to carry out your own six frame translation. Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from
Biostatistics and Bioinformatics Molecular Sequence Databases
 . 1 Description of Module Subject Name Paper Name Module Name/Title 13 03 Dr. Vijaya Khader Dr. MC Varadaraj 2 1. Objectives: In the present module, the students will learn about 1. Encoding linear sequences
. 1 Description of Module Subject Name Paper Name Module Name/Title 13 03 Dr. Vijaya Khader Dr. MC Varadaraj 2 1. Objectives: In the present module, the students will learn about 1. Encoding linear sequences
Preliminary Syllabus. Genomics. Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification
 Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Database Similarity Searching
 An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
An Introduction to Bioinformatics BSC4933/ISC5224 Florida State University Feb. 23, 2009 Database Similarity Searching Steven M. Thompson Florida State University of Department Scientific Computing How
Lecture 5: Markov models
 Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
Master s course Bioinformatics Data Analysis and Tools Lecture 5: Markov models Centre for Integrative Bioinformatics Problem in biology Data and patterns are often not clear cut When we want to make a
SAPLING: Suffix Array Piecewise Linear INdex for Genomics Michael Kirsche
 SAPLING: Suffix Array Piecewise Linear INdex for Genomics Michael Kirsche mkirsche@jhu.edu StringBio 2018 Outline Substring Search Problem Caching and Learned Data Structures Methods Results Ongoing work
SAPLING: Suffix Array Piecewise Linear INdex for Genomics Michael Kirsche mkirsche@jhu.edu StringBio 2018 Outline Substring Search Problem Caching and Learned Data Structures Methods Results Ongoing work
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A
 BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A Steve Thompson: stthompson@valdosta.edu http://www.bioinfo4u.net 1 Similarity searching and homology First, just
BIOL 7020 Special Topics Cell/Molecular: Molecular Phylogenetics. Spring 2010 Section A Steve Thompson: stthompson@valdosta.edu http://www.bioinfo4u.net 1 Similarity searching and homology First, just
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
GeneR. JORGE ARTURO ZEPEDA MARTINEZ LOPEZ HERNANDEZ JOSE FABRICIO. October 6, 2009
 GeneR JORGE ARTURO ZEPEDA MARTINEZ LOPEZ HERNANDEZ JOSE FABRICIO. jzepeda@lcg.unam.mx jlopez@lcg.unam.mx October 6, 2009 Abstract GeneR packages allow direct use of nucleotide sequences within R software.
GeneR JORGE ARTURO ZEPEDA MARTINEZ LOPEZ HERNANDEZ JOSE FABRICIO. jzepeda@lcg.unam.mx jlopez@lcg.unam.mx October 6, 2009 Abstract GeneR packages allow direct use of nucleotide sequences within R software.
BMI/CS 576 Fall 2015 Midterm Exam
 BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
Database Similarity Searching
 Database Similarity Searching Why search databases? To find out if a new DNA sequence shares similari?es with sequences already deposited in the databanks. To find proteins homologous to a puta?ve coding
Database Similarity Searching Why search databases? To find out if a new DNA sequence shares similari?es with sequences already deposited in the databanks. To find proteins homologous to a puta?ve coding
Topics of the talk. Biodatabases. Data types. Some sequence terminology...
 Topics of the talk Biodatabases Jarno Tuimala / Eija Korpelainen CSC What data are stored in biological databases? What constitutes a good database? Nucleic acid sequence databases Amino acid sequence
Topics of the talk Biodatabases Jarno Tuimala / Eija Korpelainen CSC What data are stored in biological databases? What constitutes a good database? Nucleic acid sequence databases Amino acid sequence
Jyoti Lakhani 1, Ajay Khunteta 2, Dharmesh Harwani *3 1 Poornima University, Jaipur & Maharaja Ganga Singh University, Bikaner, Rajasthan, India
 International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
International Journal of Scientific Research in Computer Science, Engineering and Information Technology 2017 IJSRCSEIT Volume 2 Issue 6 ISSN : 2456-3307 Improvisation of Global Pairwise Sequence Alignment
New generation of patent sequence databases Information Sources in Biotechnology Japan
 New generation of patent sequence databases Information Sources in Biotechnology Japan EBI is an Outstation of the European Molecular Biology Laboratory. Patent-related resources Patents Patent Resources
New generation of patent sequence databases Information Sources in Biotechnology Japan EBI is an Outstation of the European Molecular Biology Laboratory. Patent-related resources Patents Patent Resources
B L A S T! BLAST: Basic local alignment search tool. Copyright notice. February 6, Pairwise alignment: key points. Outline of tonight s lecture
 February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
Bioinformatics explained: Smith-Waterman
 Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
 Hindawi Publishing Corporation Mathematical Problems in Engineering Volume 2012, Article ID 490649, 9 pages doi:10.1155/2012/490649 Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
Hindawi Publishing Corporation Mathematical Problems in Engineering Volume 2012, Article ID 490649, 9 pages doi:10.1155/2012/490649 Research Article An Improved Scoring Matrix for Multiple Sequence Alignment
Similarity searches in biological sequence databases
 Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores.
 CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
Lab 4: Multiple Sequence Alignment (MSA)
") Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
Lab 4: Multiple Sequence Alignment (MSA) The objective of this lab is to become familiar with the features of several multiple alignment and visualization tools, including the data input and output, basic
The Effect of Inverse Document Frequency Weights on Indexed Sequence Retrieval. Kevin C. O'Kane. Department of Computer Science
 The Effect of Inverse Document Frequency Weights on Indexed Sequence Retrieval Kevin C. O'Kane Department of Computer Science The University of Northern Iowa Cedar Falls, Iowa okane@cs.uni.edu http://www.cs.uni.edu/~okane
The Effect of Inverse Document Frequency Weights on Indexed Sequence Retrieval Kevin C. O'Kane Department of Computer Science The University of Northern Iowa Cedar Falls, Iowa okane@cs.uni.edu http://www.cs.uni.edu/~okane
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Tutorial 1: Exploring the UCSC Genome Browser
 Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Trilateral Search Guidebook in Biotechnology. [Ver.1 Publication ]
![Trilateral Search Guidebook in Biotechnology. [Ver.1 Publication ]](/thumbs/89/100398076.jpg "Trilateral Search Guidebook in Biotechnology. [Ver.1 Publication ]") Trilateral Project DR2 Biotechnology Trilateral Search Guidebook in Biotechnology [Ver.1 Publication ] Part I 26 April 2007 United States Patent and trademark Office European Patent Office Japan Patent
Trilateral Project DR2 Biotechnology Trilateral Search Guidebook in Biotechnology [Ver.1 Publication ] Part I 26 April 2007 United States Patent and trademark Office European Patent Office Japan Patent
A CAM(Content Addressable Memory)-based architecture for molecular sequence matching
-based architecture for molecular sequence matching") A CAM(Content Addressable Memory)-based architecture for molecular sequence matching P.K. Lala 1 and J.P. Parkerson 2 1 Department Electrical Engineering, Texas A&M University, Texarkana, Texas, USA 2
A CAM(Content Addressable Memory)-based architecture for molecular sequence matching P.K. Lala 1 and J.P. Parkerson 2 1 Department Electrical Engineering, Texas A&M University, Texarkana, Texas, USA 2
) I R L Press Limited, Oxford, England. The protein identification resource (PIR)
 I R L Press Limited, Oxford, England. The protein identification resource (PIR)") Volume 14 Number 1 Volume 1986 Nucleic Acids Research 14 Number 1986 Nucleic Acids Research The protein identification resource (PIR) David G.George, Winona C.Barker and Lois T.Hunt National Biomedical
Volume 14 Number 1 Volume 1986 Nucleic Acids Research 14 Number 1986 Nucleic Acids Research The protein identification resource (PIR) David G.George, Winona C.Barker and Lois T.Hunt National Biomedical
Towards Declarative and Efficient Querying on Protein Structures
 Towards Declarative and Efficient Querying on Protein Structures Jignesh M. Patel University of Michigan Biology Data Types Sequences: AGCGGTA. Structure: Interaction Maps: Micro-arrays: Gene A Gene B
Towards Declarative and Efficient Querying on Protein Structures Jignesh M. Patel University of Michigan Biology Data Types Sequences: AGCGGTA. Structure: Interaction Maps: Micro-arrays: Gene A Gene B
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
Assessing Transcriptome Assembly
 Assessing Transcriptome Assembly Matt Johnson July 9, 2015 1 Introduction Now that you have assembled a transcriptome, you are probably wondering about the sequence content. Are the sequences from the
Assessing Transcriptome Assembly Matt Johnson July 9, 2015 1 Introduction Now that you have assembled a transcriptome, you are probably wondering about the sequence content. Are the sequences from the
Tutorial: How to use the Wheat TILLING database
 Tutorial: How to use the Wheat TILLING database Last Updated: 9/7/16 1. Visit http://dubcovskylab.ucdavis.edu/wheat_blast to go to the BLAST page or click on the Wheat BLAST button on the homepage. 2.
Tutorial: How to use the Wheat TILLING database Last Updated: 9/7/16 1. Visit http://dubcovskylab.ucdavis.edu/wheat_blast to go to the BLAST page or click on the Wheat BLAST button on the homepage. 2.
Sequence Alignment. part 2
 Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
CLC Server. End User USER MANUAL
 CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
CLC Server End User USER MANUAL Manual for CLC Server 10.0.1 Windows, macos and Linux March 8, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej 2 Prismet DK-8000 Aarhus C Denmark
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Introduction to Phylogenetics Week 2. Databases and Sequence Formats
 Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
Tutorial 4 BLAST Searching the CHO Genome
 Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
Tutorial 4 BLAST Searching the CHO Genome Accessing the CHO Genome BLAST Tool The CHO BLAST server can be accessed by clicking on the BLAST button on the home page or by selecting BLAST from the menu bar
In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace.
 5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
5 Multiple Match Refinement and T-Coffee In this section we describe how to extend the match refinement to the multiple case and then use T-Coffee to heuristically compute a multiple trace. This exposition
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Algorithms in Bioinformatics: A Practical Introduction. Database Search
 Algorithms in Bioinformatics: A Practical Introduction Database Search Biological databases Biological data is double in size every 15 or 16 months Increasing in number of queries: 40,000 queries per day
Algorithms in Bioinformatics: A Practical Introduction Database Search Biological databases Biological data is double in size every 15 or 16 months Increasing in number of queries: 40,000 queries per day
Sequence comparison: Local alignment
 Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
The UCSC Genome Browser
 The UCSC Genome Browser Search, retrieve and display the data that you want Materials prepared by Warren C. Lathe, Ph.D. Mary Mangan, Ph.D. www.openhelix.com Updated: Q3 2006 Version_0906 Copyright OpenHelix.
The UCSC Genome Browser Search, retrieve and display the data that you want Materials prepared by Warren C. Lathe, Ph.D. Mary Mangan, Ph.D. www.openhelix.com Updated: Q3 2006 Version_0906 Copyright OpenHelix.
Research on Pairwise Sequence Alignment Needleman-Wunsch Algorithm
 5th International Conference on Mechatronics, Materials, Chemistry and Computer Engineering (ICMMCCE 2017) Research on Pairwise Sequence Alignment Needleman-Wunsch Algorithm Xiantao Jiang1, a,*,xueliang
5th International Conference on Mechatronics, Materials, Chemistry and Computer Engineering (ICMMCCE 2017) Research on Pairwise Sequence Alignment Needleman-Wunsch Algorithm Xiantao Jiang1, a,*,xueliang
Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón
 trimal: a tool for automated alignment trimming in large-scale phylogenetics analyses Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón Version 1.2b Index of contents 1. General features
trimal: a tool for automated alignment trimming in large-scale phylogenetics analyses Salvador Capella-Gutiérrez, Jose M. Silla-Martínez and Toni Gabaldón Version 1.2b Index of contents 1. General features
Darwin: A Hardware-acceleration Framework for Genomic Sequence Alignment
 Darwin: A Hardware-acceleration Framework for Genomic Sequence Alignment Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering and Computer Science) Prof. Gill Bejerano
Darwin: A Hardware-acceleration Framework for Genomic Sequence Alignment Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering and Computer Science) Prof. Gill Bejerano
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
BGGN 213 Foundations of Bioinformatics Barry Grant
 BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
Sequencing. Computational Biology IST Ana Teresa Freitas 2011/2012. (BACs) Whole-genome shotgun sequencing Celera Genomics
 Whole-genome shotgun sequencing Celera Genomics") Computational Biology IST Ana Teresa Freitas 2011/2012 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics (BACs) 1 Must take the fragments
Computational Biology IST Ana Teresa Freitas 2011/2012 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics (BACs) 1 Must take the fragments
Important Example: Gene Sequence Matching. Corrigiendum. Central Dogma of Modern Biology. Genetics. How Nucleotides code for Amino Acids
 Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Important Example: Gene Sequence Matching Century of Biology Two views of computer science s relationship to biology: Bioinformatics: computational methods to help discover new biology from lots of data
Graph Algorithms in Bioinformatics
 Graph Algorithms in Bioinformatics Computational Biology IST Ana Teresa Freitas 2015/2016 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics
Graph Algorithms in Bioinformatics Computational Biology IST Ana Teresa Freitas 2015/2016 Sequencing Clone-by-clone shotgun sequencing Human Genome Project Whole-genome shotgun sequencing Celera Genomics
Highly Scalable and Accurate Seeds for Subsequence Alignment
 Highly Scalable and Accurate Seeds for Subsequence Alignment Abhijit Pol Tamer Kahveci Department of Computer and Information Science and Engineering, University of Florida, Gainesville, FL, USA, 32611
Highly Scalable and Accurate Seeds for Subsequence Alignment Abhijit Pol Tamer Kahveci Department of Computer and Information Science and Engineering, University of Florida, Gainesville, FL, USA, 32611
Similarity Searches on Sequence Databases
 Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
Retrieving factual data and documents using IMGT-ML in the IMGT information system
 Retrieving factual data and documents using IMGT-ML in the IMGT information system Authors : Chaume D. *, Combres K. *, Giudicelli V. *, Lefranc M.-P. * * Laboratoire d'immunogénétique Moléculaire, LIGM,
Retrieving factual data and documents using IMGT-ML in the IMGT information system Authors : Chaume D. *, Combres K. *, Giudicelli V. *, Lefranc M.-P. * * Laboratoire d'immunogénétique Moléculaire, LIGM,
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences
 BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
RESEARCH TOPIC IN BIOINFORMANTIC
 RESEARCH TOPIC IN BIOINFORMANTIC GENOME ASSEMBLY Instructor: Dr. Yufeng Wu Noted by: February 25, 2012 Genome Assembly is a kind of string sequencing problems. As we all know, the human genome is very
RESEARCH TOPIC IN BIOINFORMANTIC GENOME ASSEMBLY Instructor: Dr. Yufeng Wu Noted by: February 25, 2012 Genome Assembly is a kind of string sequencing problems. As we all know, the human genome is very