Alignments BLAST, BLAT
|
|
|
- Hannah Bruce
- 6 years ago
- Views:
Transcription
1 Alignments BLAST, BLAT

 3500Mb 100b 100000b 1 500 50000 2%")
2 Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome Linear Life cycle DNA-DNA-DNA- DNA-RNA-protein Size Amount per cell Information content 05Mb *) 3500Mb 100b b % 100% ~30%





 #")

3 The amount of genetic information in organisms Name Mycoplasma genitalium Escherichia coli Saccharomyces cerevisiae Drosophila melanogaster Caenorhabtitis Genome size (Mb) # genes elegans Homo sapiens Zea mays

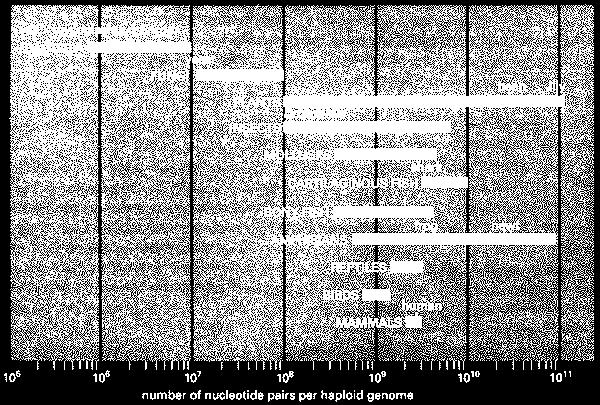

4 The amount of genetic information in organisms Largest genome: amoeba Chaos chaos (200x human genome)
5 Sequence searching - challenges Exponential growth of databases
6 Sequence searching definition Task: Query: short, new sequence (~1000 letters) Database (searching space): very many sequences Goal: find seqs homologous to the query
7 Sequence searching definition We want: fast tool primarily a filter: most sequences will be unrelated to the query fine-tune the alignment later
8 Database Search Algorithms: Sensitivity, Selectivity True Positive (TP) a homology detected (positive) correctly (true) Signal Detected Name Yes Yes True Positive No No True Negative Yes No False Negative No Yes False Positive
9 Courtesy of Gary Benson (ISSCB 2003) Database Search Algorithms: Sensitivity, Selectivity Sensitivity =TP/(TP+FN) Selectivity =TN/(TN+FP) Sensitivity Selectivity
10 What is BLAST Basic Local Alignment Search Tool Bad news: it is only a heuristics Heuristics: A rule of thumb that often helps in solving a certain class of problems, but makes no guarantees Perkins, DN (1981) The Mind's Best Work Basic idea: High scoring segments have well conserved (almost identical) part As well conserved part are identified, extend it to the real alignment - s e q - s e q u e
11 What means well conserved for BLAST? BLAST works with k-words (words of length k) k is a parameter different for DNA (>10) and proteins (24) word w 1 is T-similar to w 2 if the sum of pair scores is at least T (eg T=12) Similar 3-words W 1 : R K P W 2 : R R P Score: = 15
12 BLAST algorithm 3 basic steps 1)Preprocess 2)Scan 3)Extend 1)Preprocess the query: extract all the k-words 2)Scan for T-similar matches in database 3)Extend them to alignments
13 BLAST, Step 1: Preprocess the query 1)Preprocess 2)Scan 3)Extend Take the query (eg LVNRKPVVP) Chop it into overlapping k-words (k=3 in this case) Query: LVNRKPVVP Word1: LVN Word2: VNR Word3: NRK For each word find all similar words (scoring at least T) Eg for RKP the following 3-words are similar: QKP KKP RQP REP RRP RKP
14 Finite state machine abstract machine constant amount of memory (states) used in computation and languages recognizes regular expressions cp dmt*pdf /home/john 1)Preprocess 2)Scan 3)Extend AC*T GGC
15 BLAST, Step 2: Find exact matches 1)Preprocess 2)Scan 3)Extend with scanning Use all the T-similar k-words to build the Finite State Machine Scan for exact matches QKP KKP RQP movement REP RRP RKP VLQKPLKKPPLVKRQPCCEVVRKPLVKVIRCLA
16 BLAST, Step 3: 1)Preprocess 2)Scan 3)Extend Extending exact matches Having the list of exact matches we extend alignment in both directions Query: L V N R K P V V P T-similar: R R P Subject: G V C R R P L K C Score: till the sum of scores drops below some level X (eg X=-100) from the best known - what with gaps?
17 Gapped BLAST (now standard) 1)Preprocess 2)Scan 3)Extend gapped local alignments are computed: much, much, much slower therefore: modified Hit criteria
18 Hit criteria query pos Extends the alignment only if there are close two hits on the same diagonal sensitivity would drop without lowering T reduces extensions (90% time is spend on extensions) Gapped local alignments are computed increased sensitivity allows us raise T raising T speeds up the search close hit, same diag dbpos 1)Preprocess 2)Scan 3)Extend
19 Gapped BLAST v BLAST We end up with same speed gapped alignments! much higher sensitivity
20 BLAST flavours blastp: protein query, protein db blastn: DNA query, DNA db blastx: DNA query, protein db in all reading frames Used to find potential translation products of an unknown nucleotide sequence tblastn: protein query, DNA db database dynamically translated in all reading frames tblastx: DNA query, DNA db all translations of query against all translations of db
21 PSI-BLAST Position-Specific Iterated BLAST A profile is derived from the result of the first search Database is searched against the profile (instead of a sequence) Up to 3 iterations
22 Profile Profile is generalized form of sequence probabilities instead of a letter A C D W Y Score of the profile scorep,i, A= B p[i,b]score blosum62 A,B profile position letter
23 Constructing a profile Take significant BLAST results Make an alignment Assign weights to sequences Construct the profile A C D W Y
24 BLAT The Blast-Like Alignment Tool Large-scale genome comparison: query can be large Preprocessing phase: BLAST: query BLAT: db
25 BLAT, Step 1: Preprocess the 1)Preprocess 2)Scan 3)Extend database Index the database with k-words k=816 for nucleotides k=35 for proteins For each k-word store in which sequences it appears k-word: RKP Hashed DB: QKP: HUgn , Gene14, IG0, KKP: haemoglobin, Gene134, IG_30, RQP: HSPHOSR1, GeneA22 RKP: galactosyltransferase, IG_1 REP: haemoglobin, Gene134, IG_30, RRP: Z17368, Creatine kinase,
Preprocess")



26 Hashing associative arrays 1)Preprocess 2)Scan 3)Extend Indexing with the object Hash function: hash: small (fits in memory) possible objects - large Objects should be well spread x
27 Hashing - examples 1)Preprocess 2)Scan 3)Extend T9 Predictive Text in mobile phones hello in Multitap: 4, 4, 3, 3, 5, 5, 5, (pause) 5, 5, 5, 6, 6, 6 hello in T9: 4, 3, 5, 5, 6 Collisions: 4, 6: in, go
28 BLAT, Step 1: Index to find exact matches with hashing 1)Preprocess 2)Scan 3)Extend The database is preprocessed only once! (independent from the query) k-word: RKP Hashed DB: QKP: HUgn , Gene14, IG0, KKP: haemoglobin, Gene134, IG_30, RQP: HSPHOSR1, GeneA22 RKP: galactosyltransferase, IG_1 REP: haemoglobin, Gene134, IG_30, RRP: Z17368, Creatine kinase,
29 BLAT, Step 2: 1)Preprocess 2)Scan 3)Extend Hit criteria In a constant time we can get the sequences with a certain k-word relaxing hit definition -> improve sensitivity allow imperfect hits costly, huge hash grows a few times! shorten k (would lead to FP), but expect two hits (see BLAST)
30 BLAT, Step 3: Identifying homologous 1)Preprocess 2)Scan 3)Extend regions Exclude common k-words For all k-words from query find out the position in db For results (qpos, dbpos): split into buckets (64kbp) sort on the diagonal (diag=qposdbpos)
31 BLAT, Step 3: Identifying homologous 1)Preprocess 2)Scan 3)Extend regions continued from diagonally close hits (gap limit) create pre-clusters sort each pre-cluster on dbpos create clusters from close hits run Local Alignment for each cluster
32 Seeds improving sensitivity More general form of k-word is a seed The seed CTGTAT gives hits with both sequences CTCGTTATA CTAGTAATG
33 How to detect homology? Take the score of an maximal local alignment can it be obtained by chance? any score can be obtained from comparing (long enough) random sequences
34 What is a chance? Extracting local alignments from random sequences P-value (eg =001) The probability of obtaining the result by pure chance An alignment giving lower P-value than set by user is considered a hit
35 Best Local Alignments by chance Create random seqs, each 1000aa long Find the max local align Repeat # Alignments Score
36 The Statistics of local alignment Subst matrix must guarantee E(score(a,b)) < 0 for random a, b Analytical solution sum of iid variables -> normal distribution max of iid -> extreme value distribution (EVD)
37 Expected number of aligns E-value: the expected number of alignments scoring >= S E=K m n e S 2x size of seq -> 2x number aligns 2x S -> E drops exponentially
38 E-value depends on n, m E=K m n e S Example For comparing seqa with seqb: S=88 -> E = 0001 For comparing seqa with 1000 seqs: score 88 -> E=1 Important for db searching: n size of query, m - size of db
39 Deriving K, L E=Kmne S The above eq is theoretical result for gapless (g=inf) alignment K, L can be derived from the subst table For gapped case it seems that the equation holds we can derive K, L from experiments # Alignments Score

40 Bit score S' E=Kmne S Score S depends on the substitution table What if we want table-independent score? E=mn2 S' where S'= S ln K ln 2
41 Why does the BLAST work? Relevant riddle Are there at least 2 people in Amsterdam with the same number of hairs? At most hairs on each head people living in Amsterdam

42 Why does the BLAST work? Pigeons pigeonhole principle: having 9 boxes and 10 pigeons, there is at least one box with more than 1 pigeon n=9, k=7 case:
43 Why does the BLAST work? Average case pigeonhole principle describes the worst case! On average we'll expect two pigeons in the same box much earlier Birthday paradox: among 23 people, probability that they have the same birthday is > 05 note: 365 boxes and only 23 pigeons!
44 Birthday paradox
45 Why does the BLA[S]T work? Forget the T-similar words, now use only identities 2 sequences, 100 nucleotides each: What's the minimal sequence identity for which there's a string of 3 consecutive identities? 0 identities, 100 mismatches: 67 identities, 33 mismatches: 68st? but if seqs are 50% id, we'll detect it with prob 99% 28% id -> we'll detect it with prob 50% how is it calculated?
46 Expected sensitivity We assume that letters are independent I identity between seqs, for human-mouse 86% for DNA, 89% for proteins p word id =I k
47 Expected sensitivity Q - query size number of non-overlapping words R= Q k prob of a hit p detect =1 1 p wordid R
48 Expected specificity How many matches by chance (C)? G genome size C=Q k1 G k 1 4 k For h-m, to get 99% sensitivity we have to set k=7, and for Q=1000 C ~= 25,000,000 7h assuming 1/1000 per alignment
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Sequence Alignment. GBIO0002 Archana Bhardwaj University of Liege
 Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST - Basic Local Alignment Search Tool
 Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Lecture for ic Bioinformatics (DD2450) April 11, 2013 Searching 1. Input: Query Sequence 2. Database of sequences 3. Subject Sequence(s) 4. Output: High Segment Pairs (HSPs) Sequence Similarity Measures:
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
Introduction to Computational Molecular Biology
 18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
Lecture 5 Advanced BLAST
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
Similarity Searches on Sequence Databases
 Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
B L A S T! BLAST: Basic local alignment search tool. Copyright notice. February 6, Pairwise alignment: key points. Outline of tonight s lecture
 February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Pairwise Sequence Alignment. Zhongming Zhao, PhD
 Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
L4: Blast: Alignment Scores etc.
 L4: Blast: Alignment Scores etc. Why is Blast Fast? Silly Question Prove or Disprove: There are two people in New York City with exactly the same number of hairs. Large database search Database (n) Query
L4: Blast: Alignment Scores etc. Why is Blast Fast? Silly Question Prove or Disprove: There are two people in New York City with exactly the same number of hairs. Large database search Database (n) Query
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
MetaPhyler Usage Manual
 MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
From Smith-Waterman to BLAST
 From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores.
 CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
CS313 Exercise 4 Cover Page Fall 2017
 CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CS313 Exercise 4 Cover Page Fall 2017 Due by the start of class on Thursday, October 12, 2017. Name(s): In the TIME column, please estimate the time you spent on the parts of this exercise. Please try
CAP BLAST. BIOINFORMATICS Su-Shing Chen CISE. 8/20/2005 Su-Shing Chen, CISE 1
 CAP 5510-6 BLAST BIOINFORMATICS Su-Shing Chen CISE 8/20/2005 Su-Shing Chen, CISE 1 BLAST Basic Local Alignment Prof Search Su-Shing Chen Tool A Fast Pair-wise Alignment and Database Searching Tool 8/20/2005
CAP 5510-6 BLAST BIOINFORMATICS Su-Shing Chen CISE 8/20/2005 Su-Shing Chen, CISE 1 BLAST Basic Local Alignment Prof Search Su-Shing Chen Tool A Fast Pair-wise Alignment and Database Searching Tool 8/20/2005
Introduction to Phylogenetics Week 2. Databases and Sequence Formats
 Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
How to Run NCBI BLAST on zcluster at GACRC
 How to Run NCBI BLAST on zcluster at GACRC BLAST: Basic Local Alignment Search Tool Georgia Advanced Computing Resource Center University of Georgia Suchitra Pakala pakala@uga.edu 1 OVERVIEW What is BLAST?
How to Run NCBI BLAST on zcluster at GACRC BLAST: Basic Local Alignment Search Tool Georgia Advanced Computing Resource Center University of Georgia Suchitra Pakala pakala@uga.edu 1 OVERVIEW What is BLAST?
2) NCBI BLAST tutorial This is a users guide written by the education department at NCBI.
 NCBI BLAST tutorial This is a users guide written by the education department at NCBI.") Web resources -- Tour. page 1 of 8 This is a guided tour. Any homework is separate. In fact, this exercise is used for multiple classes and is publicly available to everyone. The entire tour will take
Web resources -- Tour. page 1 of 8 This is a guided tour. Any homework is separate. In fact, this exercise is used for multiple classes and is publicly available to everyone. The entire tour will take
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 13 G R A T I V. Iterative homology searching,
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 13 Iterative homology searching, PSI (Position Specific Iterated) BLAST basic idea use
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA
 LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA Michael Brudno, Chuong B. Do, Gregory M. Cooper, et al. Presented by Xuebei Yang About Alignments Pairwise Alignments
LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA Michael Brudno, Chuong B. Do, Gregory M. Cooper, et al. Presented by Xuebei Yang About Alignments Pairwise Alignments
BGGN 213 Foundations of Bioinformatics Barry Grant
 BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:
 FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
FastA and the chaining problem, Gunnar Klau, December 1, 2005, 10:56 4001 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem
Scoring and heuristic methods for sequence alignment CG 17
 Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Scoring and heuristic methods for sequence alignment CG 17 Amino Acid Substitution Matrices Used to score alignments. Reflect evolution of sequences. Unitary Matrix: M ij = 1 i=j { 0 o/w Genetic Code Matrix:
Similarity searches in biological sequence databases
 Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
BLAST. Basic Local Alignment Search Tool. Used to quickly compare a protein or DNA sequence to a database.
 BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
FastA & the chaining problem
 FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
FastA & the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 1 Sources for this lecture: Lectures by Volker Heun, Daniel Huson and Knut Reinert,
Lectures by Volker Heun, Daniel Huson and Knut Reinert, in particular last years lectures
 4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
4 FastA and the chaining problem We will discuss: Heuristics used by the FastA program for sequence alignment Chaining problem 4.1 Sources for this lecture Lectures by Volker Heun, Daniel Huson and Knut
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences
 BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
BIOL591: Introduction to Bioinformatics Alignment of pairs of sequences Reading in text (Mount Bioinformatics): I must confess that the treatment in Mount of sequence alignment does not seem to me a model
Single Pass, BLAST-like, Approximate String Matching on FPGAs*
 Single Pass, BLAST-like, Approximate String Matching on FPGAs* Martin Herbordt Josh Model Yongfeng Gu Bharat Sukhwani Tom VanCourt Computer Architecture and Automated Design Laboratory Department of Electrical
Single Pass, BLAST-like, Approximate String Matching on FPGAs* Martin Herbordt Josh Model Yongfeng Gu Bharat Sukhwani Tom VanCourt Computer Architecture and Automated Design Laboratory Department of Electrical
Chapter 4: Blast. Chaochun Wei Fall 2014
 Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
15-780: Graduate Artificial Intelligence. Computational biology: Sequence alignment and profile HMMs
 5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
5-78: Graduate rtificial Intelligence omputational biology: Sequence alignment and profile HMMs entral dogma DN GGGG transcription mrn UGGUUUGUG translation Protein PEPIDE 2 omparison of Different Organisms
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J.
 BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J. Buhler Prerequisites: BLAST Exercise: Detecting and Interpreting
BLAST Exercise 2: Using mrna and EST Evidence in Annotation Adapted by W. Leung and SCR Elgin from Annotation Using mrna and ESTs by Dr. J. Buhler Prerequisites: BLAST Exercise: Detecting and Interpreting
Exercise 2: Browser-Based Annotation and RNA-Seq Data
 Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Exercise 2: Browser-Based Annotation and RNA-Seq Data Jeremy Buhler July 24, 2018 This exercise continues your introduction to practical issues in comparative annotation. You ll be annotating genomic sequence
Dynamic Programming Course: A structure based flexible search method for motifs in RNA. By: Veksler, I., Ziv-Ukelson, M., Barash, D.
 Dynamic Programming Course: A structure based flexible search method for motifs in RNA By: Veksler, I., Ziv-Ukelson, M., Barash, D., Kedem, K Outline Background Motivation RNA s structure representations
Dynamic Programming Course: A structure based flexible search method for motifs in RNA By: Veksler, I., Ziv-Ukelson, M., Barash, D., Kedem, K Outline Background Motivation RNA s structure representations
Alignment of Pairs of Sequences
 Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
Bi03a_1 Unit 03a: Alignment of Pairs of Sequences Partners for alignment Bi03a_2 Protein 1 Protein 2 =amino-acid sequences (20 letter alphabeth + gap) LGPSSKQTGKGS-SRIWDN LN-ITKSAGKGAIMRLGDA -------TGKG--------
How to use KAIKObase Version 3.1.0
 How to use KAIKObase Version 3.1.0 Version3.1.0 29/Nov/2010 http://sgp2010.dna.affrc.go.jp/kaikobase/ Copyright National Institute of Agrobiological Sciences. All rights reserved. Outline 1. System overview
How to use KAIKObase Version 3.1.0 Version3.1.0 29/Nov/2010 http://sgp2010.dna.affrc.go.jp/kaikobase/ Copyright National Institute of Agrobiological Sciences. All rights reserved. Outline 1. System overview
PPI Network Alignment Advanced Topics in Computa8onal Genomics
 PPI Network Alignment 02-715 Advanced Topics in Computa8onal Genomics PPI Network Alignment Compara8ve analysis of PPI networks across different species by aligning the PPI networks Find func8onal orthologs
PPI Network Alignment 02-715 Advanced Topics in Computa8onal Genomics PPI Network Alignment Compara8ve analysis of PPI networks across different species by aligning the PPI networks Find func8onal orthologs
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
Indexing and Searching
 Indexing and Searching Introduction How to retrieval information? A simple alternative is to search the whole text sequentially Another option is to build data structures over the text (called indices)
Indexing and Searching Introduction How to retrieval information? A simple alternative is to search the whole text sequentially Another option is to build data structures over the text (called indices)
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
BIR pipeline steps and subsequent output files description STEP 1: BLAST search
 Lifeportal (Brief description) The Lifeportal at University of Oslo (https://lifeportal.uio.no) is a Galaxy based life sciences portal lifeportal.uio.no under the UiO tools section for phylogenomic analysis,
Lifeportal (Brief description) The Lifeportal at University of Oslo (https://lifeportal.uio.no) is a Galaxy based life sciences portal lifeportal.uio.no under the UiO tools section for phylogenomic analysis,
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS
 FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Sequence Alignment Heuristics
 Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Sequence Alignment Heuristics Some slides from: Iosif Vaisman, GMU mason.gmu.edu/~mmasso/binf630alignment.ppt Serafim Batzoglu, Stanford http://ai.stanford.edu/~serafim/ Geoffrey J. Barton, Oxford Protein
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Sequence Alignment: Mo1va1on and Algorithms. Lecture 2: August 23, 2012
 Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Dynamic Programming & Smith-Waterman algorithm
 m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
The BLASTER suite Documentation
 The BLASTER suite Documentation Hadi Quesneville Bioinformatics and genomics Institut Jacques Monod, Paris, France http://www.ijm.fr/ijm/recherche/equipes/bioinformatique-genomique Last modification: 05/09/06
The BLASTER suite Documentation Hadi Quesneville Bioinformatics and genomics Institut Jacques Monod, Paris, France http://www.ijm.fr/ijm/recherche/equipes/bioinformatique-genomique Last modification: 05/09/06
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Supplementary Figure 1. Fast read-mapping algorithm of BrowserGenome.
 Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Supplementary Figure 1 Fast read-mapping algorithm of BrowserGenome. (a) Indexing strategy: The genome sequence of interest is divided into non-overlapping 12-mers. A Hook table is generated that contains
Design and Evaluation of a BLAST Ungapped Extension Accelerator, Master's Thesis
 Washington University in St. Louis Washington University Open Scholarship All Computer Science and Engineering Research Computer Science and Engineering Report Number: WUCSE-2006-21 2006-01-01 Design and
Washington University in St. Louis Washington University Open Scholarship All Computer Science and Engineering Research Computer Science and Engineering Report Number: WUCSE-2006-21 2006-01-01 Design and
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment
 Multiple Sequence Alignment") CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
CISC 889 Bioinformatics (Spring 2003) Multiple Sequence Alignment Courtesy of jalview 1 Motivations Collective statistic Protein families Identification and representation of conserved sequence features
Machine Learning. Computational biology: Sequence alignment and profile HMMs
 10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
10-601 Machine Learning Computational biology: Sequence alignment and profile HMMs Central dogma DNA CCTGAGCCAACTATTGATGAA transcription mrna CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE 2 Growth
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT
 OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
Preliminary Syllabus. Genomics. Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification
 Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Preliminary Syllabus Sep 30 Oct 2 Oct 7 Oct 9 Oct 14 Oct 16 Oct 21 Oct 25 Oct 28 Nov 4 Nov 8 Introduction & Genome Assembly Sequence Comparison Gene Modeling Gene Function Identification OCTOBER BREAK
Brief review from last class
 Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
6.047 / Computational Biology: Genomes, Networks, Evolution Fall 2008
 MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT
 IADIS International Conference Applied Computing 2006 USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT Divya R. Singh Software Engineer Microsoft Corporation, Redmond, WA 98052, USA Abdullah
IADIS International Conference Applied Computing 2006 USING AN EXTENDED SUFFIX TREE TO SPEED-UP SEQUENCE ALIGNMENT Divya R. Singh Software Engineer Microsoft Corporation, Redmond, WA 98052, USA Abdullah
Searching Sequence Databases
 Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science & Engineering 2003 Searching Sequence Databases Dan E. Krane Wright State University - Main Campus,
COMPARATIVE MICROBIAL GENOMICS ANALYSIS WORKSHOP. Exercise 2: Predicting Protein-encoding Genes, BlastMatrix, BlastAtlas
 COMPARATIVE MICROBIAL GENOMICS ANALYSIS WORKSHOP Exercise 2: Predicting Protein-encoding Genes, BlastMatrix, BlastAtlas First of all connect once again to the CBS system: Open ssh shell client. Press Quick
COMPARATIVE MICROBIAL GENOMICS ANALYSIS WORKSHOP Exercise 2: Predicting Protein-encoding Genes, BlastMatrix, BlastAtlas First of all connect once again to the CBS system: Open ssh shell client. Press Quick
Optimizing multiple spaced seeds for homology search
 Optimizing multiple spaced seeds for homology search Jinbo Xu Daniel Brown Ming Li Bin Ma Abstract Optimized spaced seeds improve sensitivity and specificity in local homology search. Several authors have
Optimizing multiple spaced seeds for homology search Jinbo Xu Daniel Brown Ming Li Bin Ma Abstract Optimized spaced seeds improve sensitivity and specificity in local homology search. Several authors have
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Introduction to BLAST with Protein Sequences. Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2
 Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
Introduction to BLAST with Protein Sequences Utah State University Spring 2014 STAT 5570: Statistical Bioinformatics Notes 6.2 1 References Chapter 2 of Biological Sequence Analysis (Durbin et al., 2001)
Module: Sequence Alignment Theory and Applica8ons Session: BLAST
 Module: Sequence Alignment Theory and Applica8ons Session: BLAST Learning Objec8ves and Outcomes v Understand the principles of the BLAST algorithm v Understand the different BLAST algorithms, parameters
Module: Sequence Alignment Theory and Applica8ons Session: BLAST Learning Objec8ves and Outcomes v Understand the principles of the BLAST algorithm v Understand the different BLAST algorithms, parameters
VL Algorithmen und Datenstrukturen für Bioinformatik ( ) WS15/2016 Woche 9
 WS15/2016 Woche 9") VL Algorithmen und Datenstrukturen für Bioinformatik (19400001) WS15/2016 Woche 9 Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Contains material from
VL Algorithmen und Datenstrukturen für Bioinformatik (19400001) WS15/2016 Woche 9 Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Contains material from
Parsimony-Based Approaches to Inferring Phylogenetic Trees
 Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
Parsimony-Based Approaches to Inferring Phylogenetic Trees BMI/CS 576 www.biostat.wisc.edu/bmi576.html Mark Craven craven@biostat.wisc.edu Fall 0 Phylogenetic tree approaches! three general types! distance:
This tutorial will show you how to conduct a BLAST search. With BLAST you may:
 Gramene v. 25 Welcome to the Gramene BLAST Tutorial This tutorial will show you how to conduct a BLAST search. With BLAST you may: Search for sequence similarity matches in the Gramene database - ideal
Gramene v. 25 Welcome to the Gramene BLAST Tutorial This tutorial will show you how to conduct a BLAST search. With BLAST you may: Search for sequence similarity matches in the Gramene database - ideal
Sequence Alignment. part 2
 Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Tutorial 1: Exploring the UCSC Genome Browser
 Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.
Last updated: May 12, 2011 Tutorial 1: Exploring the UCSC Genome Browser Open the homepage of the UCSC Genome Browser at: http://genome.ucsc.edu/ In the blue bar at the top, click on the Genomes link.