DNA / RNA sequencing
|
|
|
- Lee Booker
- 5 years ago
- Views:
Transcription

1
2 Outline Ways to generate large amounts of sequence Understanding the contents of large sequence files Fasta format Fastq format Sequence quality metrics Summarizing sequence data quality/quantity Using Unix to look at large files Manipulating large files in Unix

3
4
5 DNA / RNA sequencing Sanger Long reads (800 bp), high quality targeted (primers), slow, expensive, hard to automate 454 Long reads ( bp), fairly high quality Insertions/deletions, library prep is expensive, not cheap Illumina Many, many reads, high quality Short(ish) 100bp-250bp Ion torrent error rates, throughput PacBio high error rates (10-15% errors) but very long reads (up to 100kb) Oxford nanopore
6 DNA / RNA sequencing Sanger Long reads (800 bp), high quality targeted (primers), slow, expensive, hard to automate 454 Long reads ( bp), fairly high quality Insertions/deletions, library prep is expensive, not cheap Illumina Many, many reads, high quality Short(ish) 100bp-250bp Ion torrent error rates, throughput PacBio high error rates (10-15% errors) but very long reads (up to 100kb) Oxford nanopore
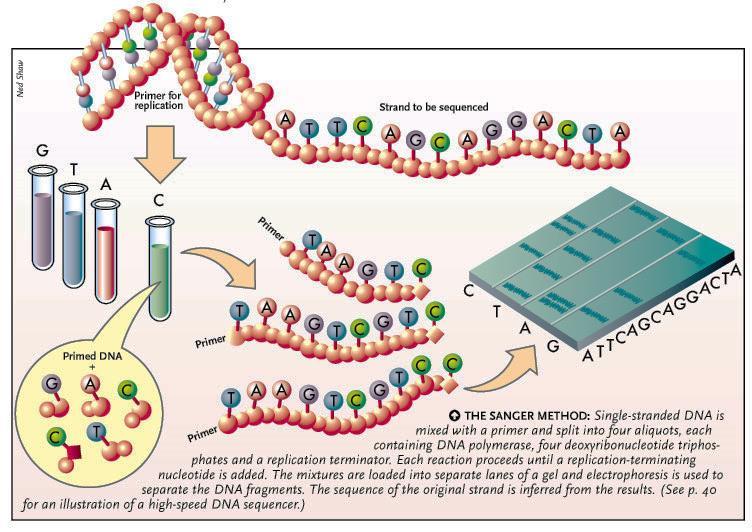
7
8 DNA / RNA sequencing Sanger Long reads (800 bp), high quality targeted (primers), slow, expensive, hard to automate 454 Long reads ( bp), fairly high quality Insertions/deletions, library prep is expensive, not cheap Illumina Many, many reads, high quality Short(ish) 100bp-250bp Ion torrent high error rates, throughput PacBio high error rates (10-15% errors) but very long reads (up to 100kb) Oxford nanopore
9
10 DNA / RNA sequencing Sanger Long reads (800 bp), high quality targeted (primers), slow, expensive, hard to automate 454 Long reads ( bp), fairly high quality Insertions/deletions, library prep is expensive, not cheap Illumina Many, many reads, high quality Short(ish) 100bp-250bp Ion torrent error rates, throughput PacBio high error rates (10-15% errors) but very long reads (up to 100kb) Oxford nanopore

11 /next-gen-fieldguide-2014/
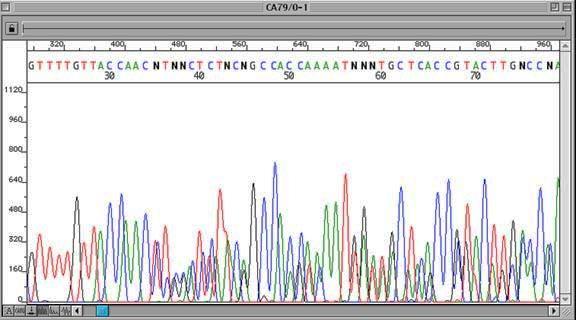
12
13 Illumina library prep Shear the DNA to specific fragment length Ligate on adaptors and barcodes
14

15
16 Illumina sequencing
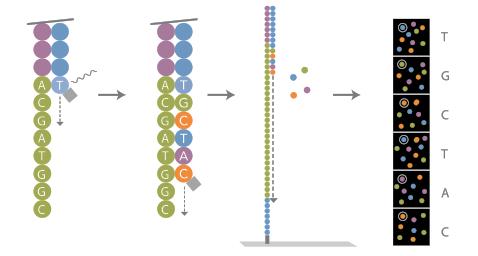
17 Illumina sequencing
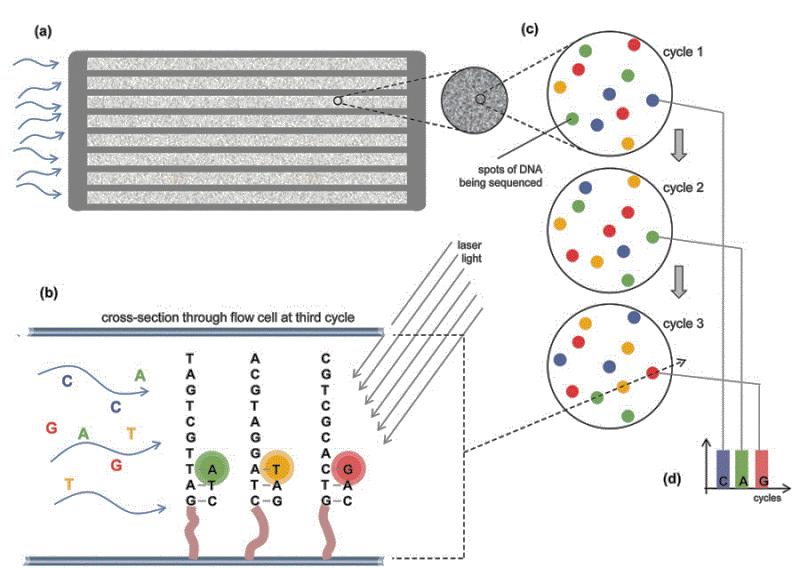
18
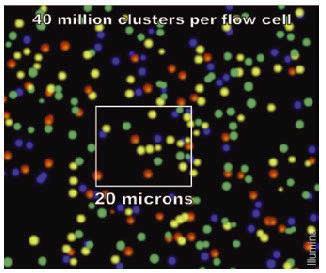
19 Illumina sequencing
20
21 Data formats Fasta Fastq.fastq.fq.fq.txt.fastq.txt SAM BAM
22 Basic Unix Unix tutorial Standard commands: pwd print working directory ls list contents of working directory cd change working directory less look at a text file man read the manual how to use a command wget get a file from another machine

23

24
25
26 An example dataset These files are already on Reference genome Arabidopsis mitochondrion wget ftp://ftp.arabidopsis.org/home/tair/sequences/mitochondrial/mitochondrial_genomic_sequence Illumina sequence for another genotype (you may have to uncompress this file)
27 Data formats Fasta Fastq.fastq.fq.fq.txt.fastq.txt SAM BAM
28 Fasta format
29 Fasta format First line: a > symbol, and a sequence name After that 1 or more lines of sequence data May have another header and other sequence after that or many headers and sequences >Cannabis sativa CBDAS mrna for cannabidiolic acid synthase, complete cds ATGAAGTGCTCAACATTCTCCTTTTGGTTTGTTTGCAAGATAATATTTTTCTTTTTCTCATTCAATATCC AAACTTCCATTGCTAATCCTCGAGAAAACTTCCTTAAATGCTTCTCGCAATATATTCCCAATAATGCAAC AAATCTAAAACTCGTATACACTCAAAACAACCCATTGTATATGTCTGTCCTAAATTCGACAATACACAAT CTTAGATTCACCTCTGACACAACCCCAAAACCACTTGTTATCGTCACTCCTTCACATGTCTCTCATATCC AAGGCACTATTCTATGCTCCAAGAAAGTTGGCTTGCAGATTCGAACTCGAAGTGGTGGTCATGATTCTGA GGGCATGTCCTACATATCTCAAGTCCCATTTGTTATAGTAGACTTGAGAAACATGCGTTCAATCAAAATA GATGTTCATAGCCAAACTGCATGGGTTGAAGCCGGAGCTACCCTTGGAGAAGTTTATTATTGGGTTAATG AGAAAAATGAGAATCTTAGTTTGGCGGCTGGGTATTGCCCTACTGTTTGCGCAGGTGGACACTTTGGTGG AGGAGGCTATGGACCATTGATGAGAAACTATGGCCTCGCGGCTGATAATATCATTGATGCACACTTAGTC
30 Now, look at the file mt.fa How do we look at this sequence? What do we know about this sequence? How do you know?
31 Data formats Fasta Fastq.fastq.fq.fq.txt.fastq.txt SAM BAM
32 Fastq format 4 line repeating pattern: 1. Header line, starting 2. DNA sequence (ATGCN) 3. spacer line, starting with + 4. Sequence quality scores
33 Looking at a fastq file using less
34 Fastq ASCII quality scores
35 Illumina quality scores Sanger format 0 to 93 using ASCII 33 to 126 Solexa/Illumina 1.0 format -5 to 62 using ASCII 59 to 126 Illumina 1.3+ format 0 to 62 using ASCII 64 to 126 Illumina and 1 are no longer used and the value 2, encoded by ASCII 66 "B", is used also at the end of reads as a Read Segment Quality Control Indicator [6].

36 Fastq ASCII quality scores

37 Looking at a fastq file using less
38 Examining the data Look at the fastq file - Less SRR fastq Please work in groups of 2-4 again, and figure out which quality score type is this?
39 Illumina quality scores Sanger format 0 to 93 using ASCII 33 to 126 Solexa/Illumina 1.0 format -5 to 62 using ASCII 59 to 126 Illumina 1.3+ format 0 to 62 using ASCII 64 to 126 Illumina and 1 are no longer used and the value 2, encoded by ASCII 66 "B", is used also at the end of reads as a Read Segment Quality Control Indicator [6].
40
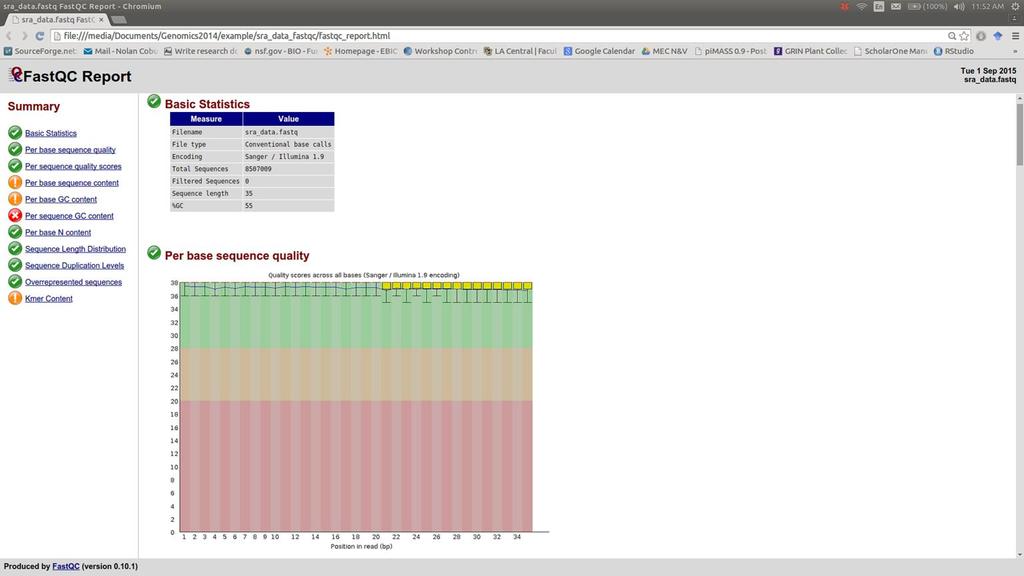
41 Evaluating quality Fastqc - a good program for quality metrics fastqc sra_data.fastq
42 For Thursday Do modules 3-4 in the tutorial Read up on fastq files:
43
44
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
Galaxy Platform For NGS Data Analyses
 Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
Galaxy Platform For NGS Data Analyses Weihong Yan wyan@chem.ucla.edu Collaboratory Web Site http://qcb.ucla.edu/collaboratory Collaboratory Workshops Workshop Outline ü Day 1 UCLA galaxy and user account
RNA-seq Data Analysis
 Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Seyed Abolfazl Motahari RNA-seq Data Analysis Basics Next Generation Sequencing Biological Samples Data Cost Data Volume Big Data Analysis in Biology تحلیل داده ها کنترل سیستمهای بیولوژیکی تشخیص بیماریها
Understanding and Pre-processing Raw Illumina Data
 Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Understanding and Pre-processing Raw Illumina Data Matt Johnson October 4, 2013 1 Understanding FASTQ files After an Illumina sequencing run, the data is stored in very large text files in a standard format
Variation among genomes
 Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
Variation among genomes Comparing genomes The reference genome http://www.ncbi.nlm.nih.gov/nuccore/26556996 Arabidopsis thaliana, a model plant Col-0 variety is from Landsberg, Germany Ler is a mutant
NGS : reads quality control
 NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
NGS : reads quality control Data used in this tutorials are available on https:/urgi.versailles.inra.fr/download/tuto/ngs-readsquality-control. Select genome solexa.fasta, illumina.fastq, solexa.fastq
Sequence Data Quality Assessment Exercises and Solutions.
 Sequence Data Quality Assessment Exercises and Solutions. Starting Note: Please do not copy and paste the commands. Characters in this document may not be copied correctly. Please type the commands and
Sequence Data Quality Assessment Exercises and Solutions. Starting Note: Please do not copy and paste the commands. Characters in this document may not be copied correctly. Please type the commands and
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy August 18, 2014 Contents 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Peter Schweitzer, Director, DNA Sequencing and Genotyping Lab
 The instruments, the runs, the QC metrics, and the output Peter Schweitzer, Director, DNA Sequencing and Genotyping Lab Overview Roche/454 GS-FLX 454 (GSRunbrowser information) Evaluating run results Errors
The instruments, the runs, the QC metrics, and the output Peter Schweitzer, Director, DNA Sequencing and Genotyping Lab Overview Roche/454 GS-FLX 454 (GSRunbrowser information) Evaluating run results Errors
Copyright 2014 Regents of the University of Minnesota
 Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Quality Control of Illumina Data using Galaxy Contents September 16, 2014 1 Introduction 2 1.1 What is Galaxy?..................................... 2 1.2 Galaxy at MSI......................................
Trimming and quality control ( )
") Trimming and quality control (2015-06-03) Alexander Jueterbock, Martin Jakt PhD course: High throughput sequencing of non-model organisms Contents 1 Overview of sequence lengths 2 2 Quality control 3 3
Trimming and quality control (2015-06-03) Alexander Jueterbock, Martin Jakt PhD course: High throughput sequencing of non-model organisms Contents 1 Overview of sequence lengths 2 2 Quality control 3 3
These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data.
 These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data. We have a few different choices for running jobs on DT2 we will explore both here. We need to alter
These will serve as a basic guideline for read prep. This assumes you have demultiplexed Illumina data. We have a few different choices for running jobs on DT2 we will explore both here. We need to alter
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
NGS Data Visualization and Exploration Using IGV
 1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
1 What is Galaxy Galaxy for Bioinformaticians Galaxy for Experimental Biologists Using Galaxy for NGS Analysis NGS Data Visualization and Exploration Using IGV 2 What is Galaxy Galaxy for Bioinformaticians
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi
 Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Colorado State University Bioinformatics Algorithms Assignment 6: Analysis of High- Throughput Biological Data Hamidreza Chitsaz, Ali Sharifi- Zarchi Although a little- bit long, this is an easy exercise
Genomics AGRY Michael Gribskov Hock 331
 Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
BIT 815: Analysis of Deep DNA Sequencing Data
 BIT 815: Analysis of Deep DNA Sequencing Data Overview: This course covers methods for analysis of data from high-throughput DNA sequencing, with or without a reference genome sequence, using free and
BIT 815: Analysis of Deep DNA Sequencing Data Overview: This course covers methods for analysis of data from high-throughput DNA sequencing, with or without a reference genome sequence, using free and
NGS NEXT GENERATION SEQUENCING
 NGS NEXT GENERATION SEQUENCING Paestum (Sa) 15-16 -17 maggio 2014 Relatore Dr Cataldo Senatore Dr.ssa Emilia Vaccaro Sanger Sequencing Reactions For given template DNA, it s like PCR except: Uses only
NGS NEXT GENERATION SEQUENCING Paestum (Sa) 15-16 -17 maggio 2014 Relatore Dr Cataldo Senatore Dr.ssa Emilia Vaccaro Sanger Sequencing Reactions For given template DNA, it s like PCR except: Uses only
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
DNA Sequencing analysis on Artemis
 DNA Sequencing analysis on Artemis Mapping and Variant Calling Tracy Chew Senior Research Bioinformatics Technical Officer Rosemarie Sadsad Informatics Services Lead Hayim Dar Informatics Technical Officer
DNA Sequencing analysis on Artemis Mapping and Variant Calling Tracy Chew Senior Research Bioinformatics Technical Officer Rosemarie Sadsad Informatics Services Lead Hayim Dar Informatics Technical Officer
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
Importing your Exeter NGS data into Galaxy:
 Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
Importing your Exeter NGS data into Galaxy: The aim of this tutorial is to show you how to import your raw Illumina FASTQ files and/or assemblies and remapping files into Galaxy. As of 1 st July 2011 Illumina
An Introduction to Linux and Bowtie
 An Introduction to Linux and Bowtie Cavan Reilly November 10, 2017 Table of contents Introduction to UNIX-like operating systems Installing programs Bowtie SAMtools Introduction to Linux In order to use
An Introduction to Linux and Bowtie Cavan Reilly November 10, 2017 Table of contents Introduction to UNIX-like operating systems Installing programs Bowtie SAMtools Introduction to Linux In order to use
MetaPhyler Usage Manual
 MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
Package Rsubread. July 21, 2013
 Package Rsubread July 21, 2013 Type Package Title Rsubread: an R package for the alignment, summarization and analyses of next-generation sequencing data Version 1.10.5 Author Wei Shi and Yang Liao with
Package Rsubread July 21, 2013 Type Package Title Rsubread: an R package for the alignment, summarization and analyses of next-generation sequencing data Version 1.10.5 Author Wei Shi and Yang Liao with
Contact: Raymond Hovey Genomics Center - SFS
 Bioinformatics Lunch Seminar (Summer 2014) Every other Friday at noon. 20-30 minutes plus discussion Informal, ask questions anytime, start discussions Content will be based on feedback Targeted at broad
Bioinformatics Lunch Seminar (Summer 2014) Every other Friday at noon. 20-30 minutes plus discussion Informal, ask questions anytime, start discussions Content will be based on feedback Targeted at broad
NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab
![NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab](/thumbs/74/70633133.jpg "NGS Sequence data. Jason Stajich. UC Riverside. jason.stajich[at]ucr.edu. twitter:hyphaltip stajichlab") NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
NGS Sequence data Jason Stajich UC Riverside jason.stajich[at]ucr.edu twitter:hyphaltip stajichlab Lecture available at http://github.com/hyphaltip/cshl_2012_ngs 1/58 NGS sequence data Quality control
README _EPGV_DataTransfer_Illumina Sequencing
 README _EPGV_DataTransfer_Illumina Sequencing I. Delivered files / Paired-ends (PE) sequences... 2 II. Flowcell (FC) Nomenclature... 2 III. Quality Control Process and EPGV Cleaning Version 1.7... 4 A.
README _EPGV_DataTransfer_Illumina Sequencing I. Delivered files / Paired-ends (PE) sequences... 2 II. Flowcell (FC) Nomenclature... 2 III. Quality Control Process and EPGV Cleaning Version 1.7... 4 A.
PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR
 PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR GOAL OF THIS SESSION Assuming that The audiences know how to perform GWAS
PRACTICAL SESSION 5 GOTCLOUD ALIGNMENT WITH BWA JAN 7 TH, 2014 STOM 2014 WORKSHOP HYUN MIN KANG UNIVERSITY OF MICHIGAN, ANN ARBOR GOAL OF THIS SESSION Assuming that The audiences know how to perform GWAS
Galaxy workshop at the Winter School Igor Makunin
 Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
Galaxy workshop at the Winter School 2016 Igor Makunin i.makunin@uq.edu.au Winter school, UQ, July 6, 2016 Plan Overview of the Genomics Virtual Lab Introduce Galaxy, a web based platform for analysis
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
de novo assembly Simon Rasmussen 36626: Next Generation Sequencing analysis DTU Bioinformatics 27626 - Next Generation Sequencing Analysis Generalized NGS analysis Data size Application Assembly: Compare
ASAP - Allele-specific alignment pipeline
 ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
ASAP - Allele-specific alignment pipeline Jan 09, 2012 (1) ASAP - Quick Reference ASAP needs a working version of Perl and is run from the command line. Furthermore, Bowtie needs to be installed on your
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Lecture 8. Sequence alignments
 Lecture 8 Sequence alignments DATA FORMATS bioawk bioawk is a program that extends awk s powerful processing of tabular data to processing tasks involving common bioinformatics formats like FASTA/FASTQ,
Lecture 8 Sequence alignments DATA FORMATS bioawk bioawk is a program that extends awk s powerful processing of tabular data to processing tasks involving common bioinformatics formats like FASTA/FASTQ,
Sep. Guide. Edico Genome Corp North Torrey Pines Court, Plaza Level, La Jolla, CA 92037
 Sep 2017 DRAGEN TM Quick Start Guide www.edicogenome.com info@edicogenome.com Edico Genome Corp. 3344 North Torrey Pines Court, Plaza Level, La Jolla, CA 92037 Notice Contents of this document and associated
Sep 2017 DRAGEN TM Quick Start Guide www.edicogenome.com info@edicogenome.com Edico Genome Corp. 3344 North Torrey Pines Court, Plaza Level, La Jolla, CA 92037 Notice Contents of this document and associated
1. Download the data from ENA and QC it:
 GenePool-External : Genome Assembly tutorial for NGS workshop 20121016 This page last changed on Oct 11, 2012 by tcezard. This is a whole genome sequencing of a E. coli from the 2011 German outbreak You
GenePool-External : Genome Assembly tutorial for NGS workshop 20121016 This page last changed on Oct 11, 2012 by tcezard. This is a whole genome sequencing of a E. coli from the 2011 German outbreak You
Data Preprocessing : Next Generation Sequencing analysis CBS - DTU Next Generation Sequencing Analysis
 Data Preprocessing 27626: Next Generation Sequencing analysis CBS - DTU Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing 27626: Next Generation Sequencing analysis CBS - DTU Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing. Next Generation Sequencing analysis DTU Bioinformatics Next Generation Sequencing Analysis
 Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Data Preprocessing Next Generation Sequencing analysis DTU Bioinformatics Generalized NGS analysis Data size Application Assembly: Compare Raw Pre- specific: Question Alignment / samples / Answer? reads
Rsubread package: high-performance read alignment, quantification and mutation discovery
 Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Quality assessment of NGS data
 Quality assessment of NGS data Ines de Santiago July 27, 2015 Contents 1 Introduction 1 2 Checking read quality with FASTQC 1 3 Preprocessing with FASTX-Toolkit 2 3.1 Preprocessing with FASTX-Toolkit:
Quality assessment of NGS data Ines de Santiago July 27, 2015 Contents 1 Introduction 1 2 Checking read quality with FASTQC 1 3 Preprocessing with FASTX-Toolkit 2 3.1 Preprocessing with FASTX-Toolkit:
Nevada Genomics Center
 Nevada Genomics Center These are general instructions on how to use dnatools to submit next generation sequencing samples to be run on either the Ion Torrent Proton or the Illumina NextSeq500. We here
Nevada Genomics Center These are general instructions on how to use dnatools to submit next generation sequencing samples to be run on either the Ion Torrent Proton or the Illumina NextSeq500. We here
Quantitative Biology Bootcamp Intro to Unix: Command Line Interface
 Quantitative Biology Bootcamp Intro to Unix: Command Line Interface Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology 2 September 2014 Running
Quantitative Biology Bootcamp Intro to Unix: Command Line Interface Frederick J Tan Bioinformatics Research Faculty Carnegie Institution of Washington, Department of Embryology 2 September 2014 Running
Pre-processing and quality control of sequence data. Barbera van Schaik KEBB - Bioinformatics Laboratory
 Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Pre-processing and quality control of sequence data Barbera van Schaik KEBB - Bioinformatics Laboratory b.d.vanschaik@amc.uva.nl Topic: quality control and prepare data for the interesting stuf Keep Throw
Rsubread package: high-performance read alignment, quantification and mutation discovery
 Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
Rsubread package: high-performance read alignment, quantification and mutation discovery Wei Shi 14 September 2015 1 Introduction This vignette provides a brief description to the Rsubread package. For
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1
 EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
EpiGnome Methyl Seq Bioinformatics User Guide Rev. 0.1 Introduction This guide contains data analysis recommendations for libraries prepared using Epicentre s EpiGnome Methyl Seq Kit, and sequenced on
The Analysis of RAD-tag Data for Association Studies
 EDEN Exchange Participant Name: Layla Freeborn Host Lab: The Kronforst Lab, The University of Chicago Dates of visit: February 15, 2013 - April 15, 2013 Title of Protocol: Rationale and Background: to
EDEN Exchange Participant Name: Layla Freeborn Host Lab: The Kronforst Lab, The University of Chicago Dates of visit: February 15, 2013 - April 15, 2013 Title of Protocol: Rationale and Background: to
2015 Workshop on Genomics. Genomics Laboratory
 2015 Workshop on Genomics Genomics Laboratory Instructors: Konrad Paszkiewicz k.h.paszkiewicz@exeter.ac.uk Objectives: By the end of the lab you will be expected to: Understand how short reads are generated.
2015 Workshop on Genomics Genomics Laboratory Instructors: Konrad Paszkiewicz k.h.paszkiewicz@exeter.ac.uk Objectives: By the end of the lab you will be expected to: Understand how short reads are generated.
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018)
") Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Darwin: A Genomic Co-processor gives up to 15,000X speedup on long read assembly (To appear in ASPLOS 2018) Yatish Turakhia EE PhD candidate Stanford University Prof. Bill Dally (Electrical Engineering
Mar. Guide. Edico Genome Inc North Torrey Pines Court, Plaza Level, La Jolla, CA 92037
 Mar 2017 DRAGEN TM Quick Start Guide www.edicogenome.com info@edicogenome.com Edico Genome Inc. 3344 North Torrey Pines Court, Plaza Level, La Jolla, CA 92037 Notice Contents of this document and associated
Mar 2017 DRAGEN TM Quick Start Guide www.edicogenome.com info@edicogenome.com Edico Genome Inc. 3344 North Torrey Pines Court, Plaza Level, La Jolla, CA 92037 Notice Contents of this document and associated
Next Generation Sequence Alignment on the BRC Cluster. Steve Newhouse 22 July 2010
 Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
Next Generation Sequence Alignment on the BRC Cluster Steve Newhouse 22 July 2010 Overview Practical guide to processing next generation sequencing data on the cluster No details on the inner workings
RASER: Reads Aligner for SNPs and Editing sites of RNA (version 0.51) Manual
 Manual") RASER: Reads Aligner for SNPs and Editing sites of RNA (version 0.51) Manual July 02, 2015 1 Index 1. System requirement and how to download RASER source code...3 2. Installation...3 3. Making index files...3
RASER: Reads Aligner for SNPs and Editing sites of RNA (version 0.51) Manual July 02, 2015 1 Index 1. System requirement and how to download RASER source code...3 2. Installation...3 3. Making index files...3
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Ensembl RNASeq Practical. Overview
 Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
Ensembl RNASeq Practical The aim of this practical session is to use BWA to align 2 lanes of Zebrafish paired end Illumina RNASeq reads to chromosome 12 of the zebrafish ZV9 assembly. We have restricted
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013
 ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
ITMO Ecole de Bioinformatique Hands-on session: smallrna-seq N. Servant 21 rd November 2013 1. Data and objectives We will use the data from GEO (GSE35368, Toedling, Servant et al. 2011). Two samples were
RNAseq analysis: SNP calling. BTI bioinformatics course, spring 2013
 RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
RNAseq analysis: SNP calling BTI bioinformatics course, spring 2013 RNAseq overview RNAseq overview Choose technology 454 Illumina SOLiD 3 rd generation (Ion Torrent, PacBio) Library types Single reads
User's Guide to DNASTAR SeqMan NGen For Windows, Macintosh and Linux
 User's Guide to DNASTAR SeqMan NGen 12.0 For Windows, Macintosh and Linux DNASTAR, Inc. 2014 Contents SeqMan NGen Overview...7 Wizard Navigation...8 Non-English Keyboards...8 Before You Begin...9 The
User's Guide to DNASTAR SeqMan NGen 12.0 For Windows, Macintosh and Linux DNASTAR, Inc. 2014 Contents SeqMan NGen Overview...7 Wizard Navigation...8 Non-English Keyboards...8 Before You Begin...9 The
Sentieon Documentation
 Sentieon Documentation Release 201808.03 Sentieon, Inc Dec 21, 2018 Sentieon Manual 1 Introduction 1 1.1 Description.............................................. 1 1.2 Benefits and Value..........................................
Sentieon Documentation Release 201808.03 Sentieon, Inc Dec 21, 2018 Sentieon Manual 1 Introduction 1 1.1 Description.............................................. 1 1.2 Benefits and Value..........................................
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Demultiplexing Illumina sequencing data containing unique molecular indexes (UMIs)
") next generation sequencing analysis guidelines Demultiplexing Illumina sequencing data containing unique molecular indexes (UMIs) See what more we can do for you at www.idtdna.com. For Research Use Only
next generation sequencing analysis guidelines Demultiplexing Illumina sequencing data containing unique molecular indexes (UMIs) See what more we can do for you at www.idtdna.com. For Research Use Only
Single/paired-end RNAseq analysis with Galaxy
 October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
October 016 Single/paired-end RNAseq analysis with Galaxy Contents: 1. Introduction. Quality control 3. Alignment 4. Normalization and read counts 5. Workflow overview 6. Sample data set to test the paired-end
Genome 373: Mapping Short Sequence Reads III. Doug Fowler
 Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Genome 373: Mapping Short Sequence Reads III Doug Fowler What is Galaxy? Galaxy is a free, open source web platform for running all sorts of computational analyses including pretty much all of the sequencing-related
Fusion Detection Using QIAseq RNAscan Panels
 Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
Fusion Detection Using QIAseq RNAscan Panels June 11, 2018 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com ts-bioinformatics@qiagen.com
SAM : Sequence Alignment/Map format. A TAB-delimited text format storing the alignment information. A header section is optional.
 Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
ls /data/atrnaseq/ egrep "(fastq fasta fq fa)\.gz" ls /data/atrnaseq/ egrep "(cn ts)[1-3]ln[^3a-za-z]\."
![ls /data/atrnaseq/ egrep (fastq fasta fq fa)\.gz ls /data/atrnaseq/ egrep (cn ts)[1-3]ln[^3a-za-z]\.](/thumbs/73/69027269.jpg "ls /data/atrnaseq/ egrep (fastq fasta fq fa)\.gz ls /data/atrnaseq/ egrep (cn ts)[1-3]ln[^3a-za-z]\.") Command line tools - bash, awk and sed We can only explore a small fraction of the capabilities of the bash shell and command-line utilities in Linux during this course. An entire course could be taught
Command line tools - bash, awk and sed We can only explore a small fraction of the capabilities of the bash shell and command-line utilities in Linux during this course. An entire course could be taught
SMRT-Portal Exercises. J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015
 SMRT-Portal Exercises J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Running SMRT-Portal in AWS see PacBio documentation We ll be running a virtual machine (VM) in the Amazon Web
SMRT-Portal Exercises J Fass UCD Genome Center Bioinformatics Core Thursday April 16, 2015 Running SMRT-Portal in AWS see PacBio documentation We ll be running a virtual machine (VM) in the Amazon Web
Mapping reads to a reference genome
 Introduction Mapping reads to a reference genome Dr. Robert Kofler October 17, 2014 Dr. Robert Kofler Mapping reads to a reference genome October 17, 2014 1 / 52 Introduction RESOURCES the lecture: http://drrobertkofler.wikispaces.com/ngsandeelecture
Introduction Mapping reads to a reference genome Dr. Robert Kofler October 17, 2014 Dr. Robert Kofler Mapping reads to a reference genome October 17, 2014 1 / 52 Introduction RESOURCES the lecture: http://drrobertkofler.wikispaces.com/ngsandeelecture
GALAXY BIOINFORMATICS WORKFLOW ENVIRONMENT. Rutger Vos, 3 April 2012
 GALAXY BIOINFORMATICS WORKFLOW ENVIRONMENT Rutger Vos, 3 April 2012 Overview Informatics in the post-genomic era The past (?) Analyses glued together using scripting languages, directly on the CLI or in
GALAXY BIOINFORMATICS WORKFLOW ENVIRONMENT Rutger Vos, 3 April 2012 Overview Informatics in the post-genomic era The past (?) Analyses glued together using scripting languages, directly on the CLI or in
2013 Workshop on Genomics, Cesky Krumlov
 2013 Workshop on Genomics, Cesky Krumlov Instructors: Part 1: Short read genomics: Remapping Konrad Paszkiewicz k.h.paszkiewicz at exeter ac uk Objectives: By the end of the workshop you will be expected
2013 Workshop on Genomics, Cesky Krumlov Instructors: Part 1: Short read genomics: Remapping Konrad Paszkiewicz k.h.paszkiewicz at exeter ac uk Objectives: By the end of the workshop you will be expected
ChIP-seq Analysis. BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute.
 ChIP-seq Analysis BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
ChIP-seq Analysis BaRC Hot Topics - March 21 st 2017 Bioinformatics and Research Computing Whitehead Institute http://barc.wi.mit.edu/hot_topics/ Outline ChIP-seq overview Experimental design Quality control/preprocessing
Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata
 Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
Analysis of RNA sequencing data sets using the Galaxy environment Dr. Gabriela Salinas Dr. Orr Shomroni Kaamini Rhaithata Microarray and Deep-sequencing core facility 30.10.2017 RNA-seq workflow I Hypothesis
ChIP-seq hands-on practical using Galaxy
 ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
ChIP-seq hands-on practical using Galaxy In this exercise we will cover some of the basic NGS analysis steps for ChIP-seq using the Galaxy framework: Quality control Mapping of reads using Bowtie2 Peak-calling
Helpful Galaxy screencasts are available at:
 This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
This user guide serves as a simplified, graphic version of the CloudMap paper for applicationoriented end-users. For more details, please see the CloudMap paper. Video versions of these user guides and
Subread/Rsubread Users Guide
 Subread/Rsubread Users Guide Rsubread v1.32.3/subread v1.6.3 25 February 2019 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
Subread/Rsubread Users Guide Rsubread v1.32.3/subread v1.6.3 25 February 2019 Wei Shi and Yang Liao Bioinformatics Division The Walter and Eliza Hall Institute of Medical Research The University of Melbourne
ChIP-seq practical: peak detection and peak annotation. Mali Salmon-Divon Remco Loos Myrto Kostadima
 ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
ChIP-seq practical: peak detection and peak annotation Mali Salmon-Divon Remco Loos Myrto Kostadima March 2012 Introduction The goal of this hands-on session is to perform some basic tasks in the analysis
Functional Genomics Research Stream. Computational Meeting: March 29, 2012 RNA-seq Analysis Pipeline
 Functional Genomics Research Stream Computational Meeting: March 29, 2012 RNA-seq Analysis Pipeline CHAPTER 2 Prepare Whole Transcriptome Libraries Fragment the whole transcriptome RNA 100 500 µg poly(a)
Functional Genomics Research Stream Computational Meeting: March 29, 2012 RNA-seq Analysis Pipeline CHAPTER 2 Prepare Whole Transcriptome Libraries Fragment the whole transcriptome RNA 100 500 µg poly(a)
Integrative Genomics Viewer. Prat Thiru
 Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
Integrative Genomics Viewer Prat Thiru 1 Overview User Interface Basics Browsing the Data Data Formats IGV Tools Demo Outline Based on ISMB 2010 Tutorial by Robinson and Thorvaldsdottir 2 Why IGV? IGV
de.nbi Nanopore Training Course Documentation Release latest
 de.nbi Nanopore Training Course Documentation Release latest Sep 21, 2018 Contents 1 The Tutorial Data Set 3 2 Basecalling 5 2.1 Basecalling with Albacore........................................ 5 2.2
de.nbi Nanopore Training Course Documentation Release latest Sep 21, 2018 Contents 1 The Tutorial Data Set 3 2 Basecalling 5 2.1 Basecalling with Albacore........................................ 5 2.2
Sequence Analysis Pipeline
 Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Sequence Analysis Pipeline Transcript fragments 1. PREPROCESSING 2. ASSEMBLY (today) Removal of contaminants, vector, adaptors, etc Put overlapping sequence together and calculate bigger sequences 3. Analysis/Annotation
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Introduction to UNIX command-line II
 Introduction to UNIX command-line II Boyce Thompson Institute 2017 Prashant Hosmani Class Content Terminal file system navigation Wildcards, shortcuts and special characters File permissions Compression
Introduction to UNIX command-line II Boyce Thompson Institute 2017 Prashant Hosmani Class Content Terminal file system navigation Wildcards, shortcuts and special characters File permissions Compression
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers
 Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Preparation of alignments for variant calling with GATK: exercise instructions for BioHPC Lab computers Data used in the exercise We will use D. melanogaster WGS paired-end Illumina data with NCBI accessions
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Quality Control of Sequencing Data
 Quality Control of Sequencing Data Surya Saha Sol Genomics Network (SGN) Boyce Thompson Institute, Ithaca, NY ss2489@cornell.edu // Twitter:@SahaSurya BTI Plant Bioinformatics Course 2017 3/27/2017 BTI
Quality Control of Sequencing Data Surya Saha Sol Genomics Network (SGN) Boyce Thompson Institute, Ithaca, NY ss2489@cornell.edu // Twitter:@SahaSurya BTI Plant Bioinformatics Course 2017 3/27/2017 BTI
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL
 QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
QIAseq DNA V3 Panel Analysis Plugin USER MANUAL User manual for QIAseq DNA V3 Panel Analysis 1.0.1 Windows, Mac OS X and Linux January 25, 2018 This software is for research purposes only. QIAGEN Aarhus
GBS Bioinformatics Pipeline(s) Overview
 Overview") GBS Bioinformatics Pipeline(s) Overview Getting from sequence files to genotypes. Pipeline Coding: Ed Buckler Jeff Glaubitz James Harriman Presentation: Terry Casstevens With supporting information from
GBS Bioinformatics Pipeline(s) Overview Getting from sequence files to genotypes. Pipeline Coding: Ed Buckler Jeff Glaubitz James Harriman Presentation: Terry Casstevens With supporting information from
Using the GBS Analysis Pipeline Tutorial
 Using the GBS Analysis Pipeline Tutorial Cornell CBSU/IGD GBS Bioinformatics Workshop September 13 & 14 2012 Step 0: If one of the CBSU BioHPC Lab workstations was reserved for you, it will be listed on
Using the GBS Analysis Pipeline Tutorial Cornell CBSU/IGD GBS Bioinformatics Workshop September 13 & 14 2012 Step 0: If one of the CBSU BioHPC Lab workstations was reserved for you, it will be listed on
Run Setup and Bioinformatic Analysis. Accel-NGS 2S MID Indexing Kits
 Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
Run Setup and Bioinformatic Analysis Accel-NGS 2S MID Indexing Kits Sequencing MID Libraries For MiSeq, HiSeq, and NextSeq instruments: Modify the config file to create a fastq for index reads Using the
replace my_user_id in the commands with your actual user ID
 Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
Exercise 1. Alignment with TOPHAT Part 1. Prepare the working directory. 1. Find out the name of the computer that has been reserved for you (https://cbsu.tc.cornell.edu/ww/machines.aspx?i=57 ). Everyone
For Research Use Only. Not for use in diagnostic procedures.
 For Research Use Only. Not for use in diagnostic procedures. P/N 101-039-100 Version 06 (October 2018) Copyright 2017-2018, Pacific Biosciences of California, Inc. All rights reserved. Information in this
For Research Use Only. Not for use in diagnostic procedures. P/N 101-039-100 Version 06 (October 2018) Copyright 2017-2018, Pacific Biosciences of California, Inc. All rights reserved. Information in this
USING BRAT-BW Table 1. Feature comparison of BRAT-bw, BRAT-large, Bismark and BS Seeker (as of on March, 2012)
") USING BRAT-BW-2.0.1 BRAT-bw is a tool for BS-seq reads mapping, i.e. mapping of bisulfite-treated sequenced reads. BRAT-bw is a part of BRAT s suit. Therefore, input and output formats for BRAT-bw are
USING BRAT-BW-2.0.1 BRAT-bw is a tool for BS-seq reads mapping, i.e. mapping of bisulfite-treated sequenced reads. BRAT-bw is a part of BRAT s suit. Therefore, input and output formats for BRAT-bw are
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching
 SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
SlopMap: a software application tool for quick and flexible identification of similar sequences using exact k-mer matching Ilya Y. Zhbannikov 1, Samuel S. Hunter 1,2, Matthew L. Settles 1,2, and James
Briefly: Bioinformatics File Formats. J Fass September 2018
 Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
Briefly: Bioinformatics File Formats J Fass September 2018 Overview ASCII Text Sequence Fasta, Fastq ~Annotation TSV, CSV, BED, GFF, GTF, VCF, SAM Binary (Data, Compressed, Executable) Data HDF5 BAM /
Cyverse tutorial 1 Logging in to Cyverse and data management. Open an Internet browser window and navigate to the Cyverse discovery environment:
 Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Cyverse tutorial 1 Logging in to Cyverse and data management Open an Internet browser window and navigate to the Cyverse discovery environment: https://de.cyverse.org/de/ Click Log in with your CyVerse
Package Rsubread. June 29, 2018
 Version 1.30.4 Date 2018-06-22 Package Rsubread June 29, 2018 Title Subread sequence alignment and counting for R Author Wei Shi and Yang Liao with contributions from Gordon K Smyth, Jenny Dai and Timothy
Version 1.30.4 Date 2018-06-22 Package Rsubread June 29, 2018 Title Subread sequence alignment and counting for R Author Wei Shi and Yang Liao with contributions from Gordon K Smyth, Jenny Dai and Timothy
HMPL User Manual. Shuying Sun or Texas State University
 HMPL User Manual Shuying Sun (ssun5211@yahoo.com or s_s355@txstate.edu), Texas State University Peng Li (pxl119@case.edu), Case Western Reserve University June 18, 2015 Contents 1. General Overview and
HMPL User Manual Shuying Sun (ssun5211@yahoo.com or s_s355@txstate.edu), Texas State University Peng Li (pxl119@case.edu), Case Western Reserve University June 18, 2015 Contents 1. General Overview and