Sequence Alignment. COMPSCI 260 Spring 2016
|
|
|
- Melvin Wright
- 5 years ago
- Views:
Transcription
1 Sequence Alignment COMPSCI 260 Spring 2016
 sequences by finding conserved regions As a step in genome assembly, and other sequence analysis tasks Understand evolugonary")
2 Why do we want to compare DNA or protein sequences? Find genes similar to known genes IdenGfy important (funcgonal) sequences by finding conserved regions As a step in genome assembly, and other sequence analysis tasks Understand evolugonary relagonships and distances (human is closer to chicken than to zebrafish) Partial CTCF protein Homologous sequences can be divided into two groups orthologous sequences: sequences that differ because they are found in different species paralogous sequences: sequences that differ because of a gene duplicagon event
3 Homology example: evolugon of globins Human α- globin and human β- globin are paralogs or orthologs? Paralogs Human α- globin and mouse α- globin are homologs or orthologs? Both
 1dtk 5pti 5pti (b) 1dtk 1dtk")

4 Homology example: sequence comparison can reveal structure 1dtk (a) 1dtk 5pti 5pti (b) 1dtk 1dtk XAKYCKLPLRIGPCKRKIPSFYYKWKAKQCLPFDYSGCGGNANRFKTIEECRRTCVG- 5pti RPDFCLEPPYTGPCKARIIRYFYNAKAGLCQTFVYGGCRAKRNNFKSAEDCMRTCGGA
5 What is the alignment problem? Input: Two protein or DNA sequences X = x 1 x 2 x m Y = y 1 y 2 y n where the x i and y i are chosen from a finite alphabet. For DNA sequences the alphabet is { A,C,G,T}. For protein sequences the alphabet is {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}. Output: the opgmal alignment of the two sequences, in the form of a list of columns of the types x i y j or x i - or - y j X = CTATGCATCA Y = GTGCACCCA CTATGCAT- CA GT--GCACC CA
6 Sequence variagons Sequences may have diverged from a common ancestor through various types of mutagons: SubsGtuGon (single nucleogde) DeleGon (single nucleogde) InserGon (single nucleogde) Inversion TransposiGon (a piece is removed and then inserted somewhere else) DuplicaGon (a piece is put in twice, or perhaps a foreign body might be insergng its genegc material into various places...) What happens if a single nucleogde dele4on or inser4on occurs in the coding porgon of the genome? x i y j Match/ mismatch x i - DeleGon (in Y rel. to X) - y j InserGon (in Y rel. to X) What happens if a single nucleogde subs4tu4on occurs in the coding porgon of the genome?
7 What is the alignment problem? Input: Two protein or DNA sequences X = x 1 x 2 x m Y = y 1 y 2 y n where the x i and y i are chosen from a finite alphabet. For DNA sequences the alphabet is { A,C,G,T}. For protein sequences the alphabet is {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}. Output: the op4mal alignment of the two sequences, in the form of a list of columns of the types x i y j or x i - or - y j We need a way to score any given alignment of X and Y
8 What is the alignment problem? Input: Two protein or DNA sequences X = x 1 x 2 x m Y = y 1 y 2 y n where the x i and y i are chosen from a finite alphabet. For DNA sequences the alphabet is { A,C,G,T}. For protein sequences the alphabet is {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}. And a funcgon Score that assigns a score to any column of the types x i y j or x i - or - y j Output: the highest scoring alignment of the two sequences, where an alignment is defined as a list of columns of the types defined above, and Score(alignment) = Σ col Score(col).
 AAAC A-GC Score(A,A) + g +")
9 Alignment scoring scheme Alignment scoring schemes reflect biological or stagsgcal observagons about the known sequences, and are frequently represented by scoring matrices Score(x i,y j ) = +1 if x i and y j is a match - > reward 1 if x i and y j is a mismatch - > penalty 2 if either x i or y j is a gap - > penalty In general, the gap penalty is denoted as g (or g) AAAC A-GC Score(A,A) + g + Score(A,G) + Score(C,C) = - 1 AAAC AGC- Score(A,A) + Score(A,G) + Score(A,C) + g = - 3?
10 Brute- force search for the opgmal alignment Given the two sequences X = x 1 x 2 x m and Y = y 1 y 2 y n we want to find the alignment that produces the best score according to the given scoring scheme. Brute- force solu4on: enumerate all the possible alignments, score each alignment, and select the alignment with the maximal score. AAAC AGC AAAC-- ---AGC AAA-C- ---AGC AAA--C ---AGC What is the total number of possible global alignments between X and Y? Idea: append n gaps to the sequence X to obtain X = x 1 x 2 x m Then we can pick n elements from X to align with the characters in Y. exponengal Gme

11 Brute- force search for the opgmal alignment Brute force: generate & score all possible alignments Time complexity: n Brute force Today s lecture , E E+58 10,000
12 OpGmal substructure property? The score is additive: for a given split (i, j), the best alignment can be computed as Best alignment of S1[1..i] and S2[1..j] + Best alignment of S1[ i+1..n] and S2[ j+1..m] Compute best alignment recursively
13 Divide and conquer? IdenGcal sub- problems! We should reuse our work!
14 SoluGon #1 MemoizaGon Create a big dicgonary (or table), indexed by aligned sequences When we encounter a new pair of sequences If it is in the dicgonary Look up the solugon If it is not in the dicgonary Compute the solugon Insert the solugon in the dicgonary Ensures that there is no duplicated work Only need to compute each sub- alignment once
15 SoluGon #2 Dynamic programming Strategy: reduce problem of best alignment of two sequences to best alignment of all prefixes of the sequences i- prefix of X is x 1 x 2 x i j- prefix of Y is y 1 y 2 y j Create a big table, indexed by (i,j) Fill it from the beginning all the way to the end The solugon is the element (n,m) Guaranteed to explore engre search space Ensures that there is no duplicated work Only need to compute each sub- alignment once! Very simply computagonally!
16 Needleman- Wunsch algorithm Consider last step in compugng the alignment of AAAC with AGC Three possible opgons; in each we will choose a different pairing (or column type) for end of alignment, and add this to best alignment of previous characters
17 Needleman- Wunsch algorithm Given an m- character sequence X, and an n- character sequence Y Construct an (m+1) (n+1) matrix F F ( i, j ) = score of the best alignment of x[1...i ] with y[1...j ]
18 Needleman- Wunsch algorithm Given an m- character sequence X, and an n- character sequence Y Construct an (m+1) (n+1) matrix F F ( i, j ) = score of the best alignment of x[1...i ] with y[1...j ] Y j X i
19 Needleman- Wunsch algorithm IniGalize first row and column of matrix Fill in rest of matrix from top to bo om, le} to right F(m, n) holds the opgmal alignment score For each F(i, j), save pointers to cells that resulted in best score Trace pointers back from F(m, n) to F(0, 0) to recover alignment Y j X i
20 IniGalizing the DP matrix
21 Global alignment example
22 Needleman- Wunsch algorithm Strategy: reduce problem of best alignment of two sequences to best alignment of all prefixes of the sequences i- prefix of X is x 1 x 2 x i j- prefix of Y is y 1 y 2 y j Matrix: F (i, j) = score of the best alignment of x 1 x 2 x i with y 1 y 2 y j
23 Needleman- Wunsch algorithm F (i, j) = score of the best alignment of x 1 x 2 x i with y 1 y 2 y j Update rule: Consider last step in compugng the alignment
24 Needleman- Wunsch algorithm IniGalize first row and column of matrix Fill in rest of matrix from top to bo om, le} to right F(m, n) holds the opgmal alignment score For each F(i, j), save pointers to cells that resulted in best score Trace pointers back from F(m, n) to F(0, 0) to recover alignment
25 Global alignment example
26 Global alignment example
27 More than one opgmal alignment? Graph search algorithms available on course website
28 Pairwise alignment via dynamic programming Works for either DNA or protein sequences, although the subsgtugon matrices are different Finds all opgmal alignments Time complexity: O(nm) Space complexity: O(nm) Can we do this faster? Yes, if we use the Four- Russians Speedup Arlazarov, Dinic, Kronrod, Faradzev (1970) Block alignment with blocks of size t = log(n) / 4 Running Gme goes from quadragc, O(n 2 ), to subquadragc: O(n 2 /logn)
29 Space complexity CompuGng the alignment requires quadragc space: O(nm) We need to keep all backtracking references in memory to reconstruct the path (backtracking)
30 Space complexity CompuGng just the score of the best alignment can be done in linear space We only need the previous column to calculate the current column, and we can then throw away that previous column once we are done using it Y X = x 1 x 2 x m Y = y 1 y 2 y n What is the space complexity? O(m) or O(n)? X How do we recover the path? We cannot. Can we compute the best alignment in linear space?
31 OpGmal global alignment in linear space The opgmal alignment must cross the middle line (i, m/2) We want to calculate the best alignment from (0,0) to (n,m) that passes through (i,m/2) where i = 0..n and represents the i- th row Define score(i) as the score of the opgmal alignment from (0,0) to (n,m) that passes through cell (i, m/2) Define (mid, m/2) as the cell where the opgmal alignment crosses the middle column score(mid) = score opgmal alignment = max 0 i n score(i)
32 OpGmal global alignment in linear space The opgmal alignment must cross the middle line (i, m/2) Prefix(i) is the score of the best alignment from (0,0) to (i,m/2) Compute Prefix(i) by DP in the le} half of the matrix Suffix(i) is the score of the best alignment from (i,m/2) to (n,m) = the score of the best alignment from (n,m) to (i,m/2) with all arrows reversed Compute Suffix(i) by DP in the right half of the reversed matrix score(i) = Prefix(i) + Suffix(i)

33 Finding the middle point

34 Finding the middle point again
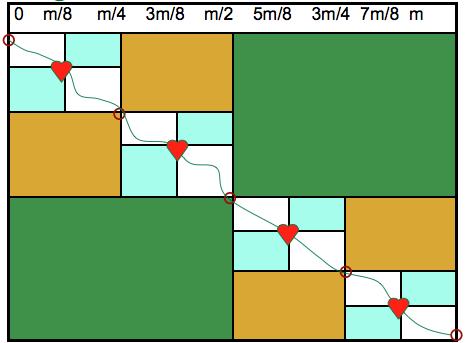
35 And again

 CompuGng")
36 Space is linear. But how about Gme complexity? On first pass, the algorithm covers the engre area nm On second pass, the algorithm covers only half of the area: nm/2 CompuGng Prefix(i) CompuGng Suffix(i)
 Geometric reducgon at each iteragon nm + nm/2 + nm/4 + < 2nm Running Gme: O(nm)")
37 Space is linear. But how about Gme complexity? On third pass, only 1/4th is covered (nm/4) Geometric reducgon at each iteragon nm + nm/2 + nm/4 + < 2nm Running Gme: O(nm) Space: O(n+m) When referring to the longest common subsequence: Hirschberg s algorithm
38 Alignment Global alignment: find best match of both sequences in their entirety Semi-global alignment: find best match without penalizing gaps on the ends of the alignment Local alignment: find best subsequence match
39 Semi- global alignment We are aligning the following sequences CAGCACTTGGATTCTCGG CAGCGTGG One possible alignment CAGCACTTGGATTCTCGG CAGC-----G-T----GG We might prefer the alignment CAGCA-CTTGGATTCTCGG ---CAGCGTGG We need a new scoring scheme Score: 8(1)+0(- 1)+10(- 2) = - 12 Score: 6(1)+1(- 1)+12(- 2) = - 19 IntuiGvely, we do not penalize missing ends of the sequence We would like to model this intuigon
40 Semi- global alignment IniGalize first row and column according to gap penalty Fill in the matrix Global alignment Report F(m,n) as the opgmal score Semi- global alignment IniGalize first row and column with 0 Fill in the matrix, as above Report max over last row and last column as the opgmal score

41 Semi- global alignment IniGalize first row and column according to gap penalty Fill in the matrix Global alignment Report F(m,n) as the opgmal score Semi- global alignment IniGalize first row and column with 0 Fill in the matrix, as above Report max over last row and last column as the opgmal score
42 Semi- global alignment One possible alignment CAGCACTTGGATTCTCGG CAGC-----G-T----GG We might prefer the alignment CAGCA-CTTGGATTCTCGG Score: 8(1)+0(- 1)+10(- 2) = CAGCGTGG Score: 6(1)+1(- 1)+12(- 2) = - 19 With the new scoring scheme (semi- global alignment) CAGCA-CTTGGATTCTCGG ---CAGCGTGG Score: 6(1)+1(- 1)+1(- 2)+11(- 0) = 3
43 Semi- global alignments ApplicaGons: Finding a gene in a genome Aligning a read onto an assembly Finding the best alignment of a PCR primer query target These situagons have in common One sequence is much shorter than the other Alignment should span the engre length of the smaller sequence No need to align the engre length of the longer sequence Local sequence alignment: best alignment between subsequences of X and Y
44 Local alignment problem: definigon Input: Two protein or DNA sequences X = x 1 x 2 x m Y = y 1 y 2 y n where the x i and y i are chosen from a finite alphabet. For DNA sequences the alphabet is { A,C,G,T}. For protein sequences the alphabet is {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}. And a funcgon Score that assigns a score to any column of the types x i y j or x i - or - y j Output: the highest scoring alignment between subsequences of X and Y, where an alignment is defined as a list of columns of the types defined above, and Score(alignment) = Σ col Score(col).
45 Local alignment Local alignment is much more common than global alignment Example: aligning two protein sequences that have a common domain but are otherwise different Compared to global alignment, the local alignment problem appears to be significantly more complex Naïve approach: given that we know how to compute the global alignment between two sequences in O(mn) Gme we can take all possible combinagons of substrings of X and substrings of Y, and run Needleman- Wunsch Smith-Waterman algorithm - Smith, T.F. & Waterman, M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol. 147: Running Gme? O(m 3 n 3 ) We can improve this significantly by using a DP approach
46 Global vs. local alignment Global alignment Y F (i, j) = score of the best alignment of x[1...i ] with y[1...j ] X Local alignment If the best alignment up to some point has a negagve score, it is be er to start a new alignment (score 0)
47 Global vs. local alignment Global alignment Y F (i, j) = score of the best alignment of x[1...i ] with y[1...j ] X Local alignment F(i, j) = score of the best If alignment the best alignment of a suffix up of to x[1...i some ] point has a negagve score, it is be er with a suffix of y[1...j ] to start a new alignment (score 0)
48 Local alignment Smith- Waterman algorithm Update rule: F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] Ini4aliza4on? Previously (global alignment) we inigalized with gap penalges Now (local alignment) we inigalize the first row and column with 0
49 Local alignment Smith- Waterman algorithm Update rule: F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] Op4mal alignment score? Global: F(m,n) Semi- global: maximum over last row F(m,j) and column F(i,n) Local:???
50 Local alignment Smith- Waterman algorithm Update rule: F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] Op4mal alignment score? Global: F(m,n) Semi- global: maximum over last row F(m,j) and column F(i,n) Local: maximum F(i,j) over all i=1..m and j=1..n
51 Local alignment Smith- Waterman algorithm Update rule: F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] Ini4aliza4on: we inigalize the first row and column with 0 Op4mal alignment score: maximum F(i,j) over all i=1..m and j=1..n Traceback: Start from the cell containing the opgmal score Stop when we reach the value 0
52 Local alignment example
53 Local alignment example
54 Local alignment Smith- Waterman algorithm Time complexity: O(mn) Space complexity: O(mn), can be brought to O(m+n) Is the solugon opgmal? Have we searched the engre space of alignments between substrings of X and Y? Yes. Because a substring of a string is the suffix of a prefix. F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] SoluGon: maximum F(i,j) over all i=1..m and j=1..n MulGple opgmal alignments
 = score of the best alignment of a suffix of x[1.")
55 Local alignment Smith- Waterman algorithm Time complexity: O(mn) Space complexity: O(mn), can be brought to O(m+n) Is the solugon opgmal? Have we searched the engre space of alignments between substrings of X and Y? Yes. Because a substring of a string is the suffix of a prefix. F(i, j) = score of the best alignment of a suffix of x[1...i ] with a suffix of y[1...j ] SoluGon: maximum F(i,j) over all i=1..m and j=1..n MulGple opgmal alignments
56 Global and local alignment Global alignment F(i, j) = score of the best alignment of x[1...i ] with y[1...j ] Match/mismatch score Local alignment Gap penalty
57 Scoring matches/mismatches - revisited Alignment scoring schemes reflect biological or stagsgcal observagons about the known sequences, and are frequently represented by scoring matrices Purines Guanine (G) Adenine (A) Pyrimidines Cytosine (C) Thymine (T)
58 Scoring matches/mismatches - revisited DNA mutagons Transi4ons: subsgtugons that occur between the two- ring purines (A G and G A) or between the one- ring pyrimidines (C T and T C). Because these subsgtugons do not require a change in the number of rings, they occur more frequently than the other subsgtugons. Transversions: slower- rate subsgtugons that change a purine to a pyrimidine or vice versa (A C, A T, G C, and G T). Purines Guanine (G) Adenine (A) Pyrimidines Cytosine (C) Thymine (T)
59 Scoring matches/mismatches - revisited For protein sequence alignment, some amino acids have similar structures and can be more easily subsgtuted in nature Glutamic acid (D) Aspartic acid (D) Tryptophan (W)
60 Chemical structures of the 20 common amino acids
61 Protein subsgtugon matrices A biologist with good intuigon could come up with a decent scoring scheme (invent 210 scores) But we would like to have some theory behind these scores, that reflects the similarity between the residues SubsGtuGon scores can be derived from probabilisgc models of evolugon Durbin et al. Biological Sequence Analysis
62 SubsGtuGon matrices for protein sequences Two popular sets of matrices for protein sequences PAM matrices [Dayhoff et al., 1978] BLOSUM matrices [Henikoff & Henikoff, 1992] Both try to capture the relagve subsgtutability of amino acid pairs in the context of evolugon Point Accepted MutaGon 1 PAM unit = PAM 1 = one mutagon per 100 amino acids A}er 100 PAMs of evolugon, not every residue will have changed some residues may have mutated several Gmes some residues may have returned to their original state some residues may not have changed at all
63 SubsGtuGon matrices for protein sequences PAM250 is a widely used matrix:
64 SubsGtuGon matrices for protein sequences BLOSUM Blocks SubsGtuGon Matrix Scores derived from observaeons of the frequencies of subsgtugons in blocks of local alignments in related proteins Accounts for evolugonarily divergent sequences Matrix name indicates evolugonary distance BLOSUM62 was created using sequences sharing no more than 62% idengty
65 SubsGtuGon matrices for protein sequences BLOSUM50 Common aminoacids have low weights Positive for chemically similar amino-acids Rare amino-acids have high weights
66 Global and local alignment Global alignment F(i, j) = score of the best alignment of x[1...i ] with y[1...j ] Match/mismatch score Local alignment Gap penalty

67 Gap penalges vs. These have the same score But the second one is o}en more plausible A single insergon of GAAT into the first string could change it into the second Current scoring scheme assumes the gaps occurred independently
68 Gap penalges Current scoring scheme: the cost associated with a gap of length k is g*k This is a linear gap penalty We would like to have a gap penalty funcgon where long gaps are not penalized that heavily OpGon: penalty for opening a gap: h penalty for extending a gap: g < h The cost associated with a gap of length k: This is an affine gap penalty
69 Linear vs. affine gap penalty Linear gap: Affine gap: Y j F(i, j) = score of the best alignment of x[1...i ] with y[1...j ] X i
70 Linear vs. affine gap penalty Linear gap: Affine gap: Y j Y j X X i i
71 Linear vs. affine gap penalty Linear gap: Affine gap: Y j " $ $ $ $ F(i, j) = max # $ $ $ $ % F(i 1, j 1) + s(x i, y j ) F(i 1, j) + w(1) F(i 2, j) + w(2)... F(i, j 1) + w(1) F(i, j 2) + w(2)... X i
72 DP for the affine gap penalty case? Affine gap: Y j " $ $ $ $ F(i, j) = max # $ $ $ $ % F(i 1, j 1) + s(x i, y j ) F(i 1, j) + w(1) F(i 2, j) + w(2)... F(i, j 1) + w(1) F(i, j 2) + w(2)... X i We can use the same approach we used for the linear gap, but Running Gme increases from O(mn) to O(mn(m+n)) For m=n, the increase is from O(n 2 ) to O(n 3 )
73 DP for the affine gap penalty case We can reduce the Gme to O(mn), but we need 3 matrices instead of 1 M(i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to y[j] x i y j I x (i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to a gap I y (i,j) = score of the best alignment of x[1...i] with y[1...j] given that y[j] is aligned to a gap x i - - y j
74 DP for the affine gap penalty case x i y j x i - " $ M (i, j) = max # $ % $ " $ I x (i, j) = max # $ % $ M (i 1, j 1) + s(x i, y j ) I x (i 1, j 1) + s(x i, y j ) I y (i 1, j 1) + s(x i, y j ) M (i 1, j) + h + g I x (i 1, j) + g I y (i 1, j) + h + g - y j " $ I y (i, j) = max # $ % $ M (i, j 1) + h + g I x (i, j 1) + h + g I y (i, j 1) + g
75 DP for the affine gap penalty case - inigalizagon M(i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to y[j] I x (i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to a gap I y (i,j) = score of the best alignment of x[1...i] with y[1...j] given that y[j] is aligned to a gap x i y j x i - - y j M(0,0) = 0 M(0,j) = the score of best alignment between 0 characters of X and j characters of Y that ends in a match = - because no such alignment can exist = M(i,0) I x (i,0) = h + g*i I y (0,j) = h + g*j I x (0,j) = - I y (i,0) = -
76 DP for the affine gap penalty case - traceback M(i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to y[j] I x (i,j) = score of the best alignment of x[1...i] with y[1...j] given that x[i] is aligned to a gap I y (i,j) = score of the best alignment of x[1...i] with y[1...j] given that y[j] is aligned to a gap x i y j x i - - y j Start at largest of M(m,n), I x (m,n), I y (m,n) Stop at any of M(0,0), I x (0,0), I y (0,0) Note that pointers may traverse all three matrices

77 3- leveled Manha an grid
78 Global alignment example affine gap penalty
79 Global alignment example affine gap penalty
80 DP for the affine gap penalty case Global alignment x i y j x i - - y j " $ M (i, j) = max # $ % $ " $ I x (i, j) = max # $ % $ " $ I y (i, j) = max # $ % $ M (i 1, j 1) + s(x i, y j ) I x (i 1, j 1) + s(x i, y j ) I y (i 1, j 1) + s(x i, y j ) M (i 1, j) + h + g I x (i 1, j) + g I y (i 1, j) + h + g M (i, j 1) + h + g I x (i, j 1) + h + g I y (i, j 1) + g
81 DP for the affine gap penalty case x i y j Local alignment " M (i 1, j 1) + s(x i, y j ) $ $ I M (i, j) = max x (i 1, j 1) + s(x i, y j ) # $ I y (i 1, j 1) + s(x i, y j ) $ % 0 x i - - y j " $ I x (i, j) = max # $ % $ " $ I y (i, j) = max # $ % $ M (i 1, j) + h + g I x (i 1, j) + g I y (i 1, j) + h + g M (i, j 1) + h + g I x (i, j 1) + h + g I y (i, j 1) + g
82 DP for the affine gap penalty case IniGalizaGon Global alignment M(0,0) = 0 M(i,0) = M(0,j) = - I x (i,0) = h + g*i I y (0,j) = h + g*j Local alignment M(0,0) = 0 M(i,0) = M(0,j) = 0 I x (i,0) = 0 I y (0,j) = 0 Traceback Start at largest of: M(m,n), I x (m,n), I y (m,n) Stop at any of: M(0,0), I x (0,0), I y (0,0) Start at largest M(i,j) Stop at any M(i,j)=0
83 DP for the affine gap penalty case Complexity Fill in thee m x n matrices - > 3mn subproblems Each one takes constant Gme Total rungme O(mn) Space complexity O(mn)
84 Gap penalty funcgons Linear gap penalty: Time: O(n 2 ) Space: O(n 2 ) Affine gap penalty: Time: O(n 2 ) Space: O(n 2 ) h Concave gap penalty funcgon: Time: O(n 3 ) Space: O(n 2 )
85 Pairwise sequence alignment What type of alignment should we consider? (global, semi- global, local) Can be done in O(nm) Gme using dynamic programming, with either linear or affine gap penalges Can be done in O(nm) space, or even O(n+m) space We can find all alignments that have the best score Works for either DNA or protein sequences, with different subsgtugon matrices
Lecture 10. Sequence alignments
 Lecture 10 Sequence alignments Alignment algorithms: Overview Given a scoring system, we need to have an algorithm for finding an optimal alignment for a pair of sequences. We want to maximize the score
Lecture 10 Sequence alignments Alignment algorithms: Overview Given a scoring system, we need to have an algorithm for finding an optimal alignment for a pair of sequences. We want to maximize the score
Pairwise Sequence Alignment: Dynamic Programming Algorithms. COMP Spring 2015 Luay Nakhleh, Rice University
 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 - Spring 2015 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment The number of all possible pairwise alignments (if
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh
 Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh Overlap detection: Semi-Global Alignment An overlap of two sequences is considered an
Computational Biology Lecture 4: Overlap detection, Local Alignment, Space Efficient Needleman-Wunsch Saad Mneimneh Overlap detection: Semi-Global Alignment An overlap of two sequences is considered an
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD
 Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University
 1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
1 Pairwise Sequence Alignment: Dynamic Programming Algorithms COMP 571 Luay Nakhleh, Rice University DP Algorithms for Pairwise Alignment 2 The number of all possible pairwise alignments (if gaps are allowed)
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST
 An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
An Analysis of Pairwise Sequence Alignment Algorithm Complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and Gapped BLAST Alexander Chan 5075504 Biochemistry 218 Final Project An Analysis of Pairwise
Lecture 12: Divide and Conquer Algorithms
 Lecture 12: Divide and Conquer Algorithms Study Chapter 7.1 7.4 1 Divide and Conquer Algorithms Divide problem into sub-problems Conquer by solving sub-problems recursively. If the sub-problems are small
Lecture 12: Divide and Conquer Algorithms Study Chapter 7.1 7.4 1 Divide and Conquer Algorithms Divide problem into sub-problems Conquer by solving sub-problems recursively. If the sub-problems are small
Divide and Conquer Algorithms. Problem Set #3 is graded Problem Set #4 due on Thursday
 Divide and Conquer Algorithms Problem Set #3 is graded Problem Set #4 due on Thursday 1 The Essence of Divide and Conquer Divide problem into sub-problems Conquer by solving sub-problems recursively. If
Divide and Conquer Algorithms Problem Set #3 is graded Problem Set #4 due on Thursday 1 The Essence of Divide and Conquer Divide problem into sub-problems Conquer by solving sub-problems recursively. If
Algorithmic Approaches for Biological Data, Lecture #20
 Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Mouse, Human, Chimpanzee
 More Alignments 1 Mouse, Human, Chimpanzee Mouse to Human Chimpanzee to Human 2 Mouse v.s. Human Chromosome X of Mouse to Human 3 Local Alignment Given: two sequences S and T Find: substrings of S and
More Alignments 1 Mouse, Human, Chimpanzee Mouse to Human Chimpanzee to Human 2 Mouse v.s. Human Chromosome X of Mouse to Human 3 Local Alignment Given: two sequences S and T Find: substrings of S and
Lecture 3: February Local Alignment: The Smith-Waterman Algorithm
 CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
CSCI1820: Sequence Alignment Spring 2017 Lecture 3: February 7 Lecturer: Sorin Istrail Scribe: Pranavan Chanthrakumar Note: LaTeX template courtesy of UC Berkeley EECS dept. Notes are also adapted from
Brief review from last class
 Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Sequence Alignment Brief review from last class DNA is has direction, we will use only one (5 -> 3 ) and generate the opposite strand as needed. DNA is a 3D object (see lecture 1) but we will model it
Divide and Conquer. Bioinformatics: Issues and Algorithms. CSE Fall 2007 Lecture 12
 Divide and Conquer Bioinformatics: Issues and Algorithms CSE 308-408 Fall 007 Lecture 1 Lopresti Fall 007 Lecture 1-1 - Outline MergeSort Finding mid-point in alignment matrix in linear space Linear space
Divide and Conquer Bioinformatics: Issues and Algorithms CSE 308-408 Fall 007 Lecture 1 Lopresti Fall 007 Lecture 1-1 - Outline MergeSort Finding mid-point in alignment matrix in linear space Linear space
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it.
 Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Sequence Alignments Overview Sequence alignment is an essential concept for bioinformatics, as most of our data analysis and interpretation techniques make use of it. Sequence alignment means arranging
Divide & Conquer Algorithms
 Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Divide & Conquer Algorithms
 Divide & Conquer Algorithms Outline 1. MergeSort 2. Finding the middle vertex 3. Linear space sequence alignment 4. Block alignment 5. Four-Russians speedup 6. LCS in sub-quadratic time Section 1: MergeSort
Divide & Conquer Algorithms Outline 1. MergeSort 2. Finding the middle vertex 3. Linear space sequence alignment 4. Block alignment 5. Four-Russians speedup 6. LCS in sub-quadratic time Section 1: MergeSort
Dynamic Programming: Sequence alignment. CS 466 Saurabh Sinha
 Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Dynamic Programming: Sequence alignment CS 466 Saurabh Sinha DNA Sequence Comparison: First Success Story Finding sequence similarities with genes of known function is a common approach to infer a newly
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Today s Lecture. Edit graph & alignment algorithms. Local vs global Computational complexity of pairwise alignment Multiple sequence alignment
 Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
Today s Lecture Edit graph & alignment algorithms Smith-Waterman algorithm Needleman-Wunsch algorithm Local vs global Computational complexity of pairwise alignment Multiple sequence alignment 1 Sequence
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties
 Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
Sequence analysis Pairwise sequence alignment
 UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
UMF11 Introduction to bioinformatics, 25 Sequence analysis Pairwise sequence alignment 1. Sequence alignment Lecturer: Marina lexandersson 12 September, 25 here are two types of sequence alignments, global
Local Alignment & Gap Penalties CMSC 423
 Local Alignment & ap Penalties CMSC 423 lobal, Semi-global, Local Alignments Last time, we saw a dynamic programming algorithm for global alignment: both strings s and t must be completely matched: s t
Local Alignment & ap Penalties CMSC 423 lobal, Semi-global, Local Alignments Last time, we saw a dynamic programming algorithm for global alignment: both strings s and t must be completely matched: s t
Divide & Conquer Algorithms
 Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Divide & Conquer Algorithms Outline MergeSort Finding the middle point in the alignment matrix in linear space Linear space sequence alignment Block Alignment Four-Russians speedup Constructing LCS in
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
EECS 4425: Introductory Computational Bioinformatics Fall Suprakash Datta
 EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
Acceleration of Algorithm of Smith-Waterman Using Recursive Variable Expansion.
 www.ijarcet.org 54 Acceleration of Algorithm of Smith-Waterman Using Recursive Variable Expansion. Hassan Kehinde Bello and Kazeem Alagbe Gbolagade Abstract Biological sequence alignment is becoming popular
www.ijarcet.org 54 Acceleration of Algorithm of Smith-Waterman Using Recursive Variable Expansion. Hassan Kehinde Bello and Kazeem Alagbe Gbolagade Abstract Biological sequence alignment is becoming popular
Sequence Alignment 1
 Sequence Alignment 1 Nucleotide and Base Pairs Purine: A and G Pyrimidine: T and C 2 DNA 3 For this course DNA is double-helical, with two complementary strands. Complementary bases: Adenine (A) - Thymine
Sequence Alignment 1 Nucleotide and Base Pairs Purine: A and G Pyrimidine: T and C 2 DNA 3 For this course DNA is double-helical, with two complementary strands. Complementary bases: Adenine (A) - Thymine
6.047 / Computational Biology: Genomes, Networks, Evolution Fall 2008
 MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
CMSC423: Bioinformatic Algorithms, Databases and Tools Lecture 8. Note
 MS: Bioinformatic lgorithms, Databases and ools Lecture 8 Sequence alignment: inexact alignment dynamic programming, gapped alignment Note Lecture 7 suffix trees and suffix arrays will be rescheduled Exact
MS: Bioinformatic lgorithms, Databases and ools Lecture 8 Sequence alignment: inexact alignment dynamic programming, gapped alignment Note Lecture 7 suffix trees and suffix arrays will be rescheduled Exact
Special course in Computer Science: Advanced Text Algorithms
 Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Special course in Computer Science: Advanced Text Algorithms Lecture 6: Alignments Elena Czeizler and Ion Petre Department of IT, Abo Akademi Computational Biomodelling Laboratory http://www.users.abo.fi/ipetre/textalg
Notes on Dynamic-Programming Sequence Alignment
 Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Notes on Dynamic-Programming Sequence Alignment Introduction. Following its introduction by Needleman and Wunsch (1970), dynamic programming has become the method of choice for rigorous alignment of DNA
Dynamic Programming Part I: Examples. Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, / 77
 Dynamic Programming -class4- July 25th, / 77") Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Dynamic Programming Part I: Examples Bioinfo I (Institut Pasteur de Montevideo) Dynamic Programming -class4- July 25th, 2011 1 / 77 Dynamic Programming Recall: the Change Problem Other problems: Manhattan
Dynamic Programming Comp 122, Fall 2004
 Dynamic Programming Comp 122, Fall 2004 Review: the previous lecture Principles of dynamic programming: optimization problems, optimal substructure property, overlapping subproblems, trade space for time,
Dynamic Programming Comp 122, Fall 2004 Review: the previous lecture Principles of dynamic programming: optimization problems, optimal substructure property, overlapping subproblems, trade space for time,
Programming assignment for the course Sequence Analysis (2006)
") Programming assignment for the course Sequence Analysis (2006) Original text by John W. Romein, adapted by Bart van Houte (bart@cs.vu.nl) Introduction Please note: This assignment is only obligatory for
Programming assignment for the course Sequence Analysis (2006) Original text by John W. Romein, adapted by Bart van Houte (bart@cs.vu.nl) Introduction Please note: This assignment is only obligatory for
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Lecture 9: Core String Edits and Alignments
 Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Biosequence Algorithms, Spring 2005 Lecture 9: Core String Edits and Alignments Pekka Kilpeläinen University of Kuopio Department of Computer Science BSA Lecture 9: String Edits and Alignments p.1/30 III:
Dynamic Programming & Smith-Waterman algorithm
 m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
BLAST. Basic Local Alignment Search Tool. Used to quickly compare a protein or DNA sequence to a database.
 BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
BLAST Basic Local Alignment Search Tool Used to quickly compare a protein or DNA sequence to a database. There is no such thing as a free lunch BLAST is fast and highly sensitive compared to competitors.
Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--
 Sequence Alignment Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC-- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Distance from sequences
Sequence Alignment Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC-- TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Distance from sequences
Bioinformatics explained: Smith-Waterman
 Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Bioinformatics Explained Bioinformatics explained: Smith-Waterman May 1, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com
Sequence Comparison: Dynamic Programming. Genome 373 Genomic Informatics Elhanan Borenstein
 Sequence omparison: Dynamic Programming Genome 373 Genomic Informatics Elhanan Borenstein quick review: hallenges Find the best global alignment of two sequences Find the best global alignment of multiple
Sequence omparison: Dynamic Programming Genome 373 Genomic Informatics Elhanan Borenstein quick review: hallenges Find the best global alignment of two sequences Find the best global alignment of multiple
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence comparison: Local alignment
 Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
Alignment ABC. Most slides are modified from Serafim s lectures
 Alignment ABC Most slides are modified from Serafim s lectures Complete genomes Evolution Evolution at the DNA level C ACGGTGCAGTCACCA ACGTTGCAGTCCACCA SEQUENCE EDITS REARRANGEMENTS Sequence conservation
Alignment ABC Most slides are modified from Serafim s lectures Complete genomes Evolution Evolution at the DNA level C ACGGTGCAGTCACCA ACGTTGCAGTCCACCA SEQUENCE EDITS REARRANGEMENTS Sequence conservation
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Outline. Sequence Alignment. Types of Sequence Alignment. Genomics & Computational Biology. Section 2. How Computers Store Information
 enomics & omputational Biology Section Lan Zhang Sep. th, Outline How omputers Store Information Sequence lignment Dot Matrix nalysis Dynamic programming lobal: NeedlemanWunsch lgorithm Local: SmithWaterman
enomics & omputational Biology Section Lan Zhang Sep. th, Outline How omputers Store Information Sequence lignment Dot Matrix nalysis Dynamic programming lobal: NeedlemanWunsch lgorithm Local: SmithWaterman
Sequence Alignment. part 2
 Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Sequence Alignment part 2 Dynamic programming with more realistic scoring scheme Using the same initial sequences, we ll look at a dynamic programming example with a scoring scheme that selects for matches
Alignment of Long Sequences
 Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Alignment of Long Sequences BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2009 Mark Craven craven@biostat.wisc.edu Pairwise Whole Genome Alignment: Task Definition Given a pair of genomes (or other large-scale
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Divya R. Singh. Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques. February A Thesis Presented by
 Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques A Thesis Presented by Divya R. Singh to The Faculty of the Graduate College of the University of Vermont In Partial Fulfillment of
Faster Sequence Alignment using Suffix Tree and Data-Mining Techniques A Thesis Presented by Divya R. Singh to The Faculty of the Graduate College of the University of Vermont In Partial Fulfillment of
Mapping Reads to Reference Genome
 Mapping Reads to Reference Genome DNA carries genetic information DNA is a double helix of two complementary strands formed by four nucleotides (bases): Adenine, Cytosine, Guanine and Thymine 2 of 31 Gene
Mapping Reads to Reference Genome DNA carries genetic information DNA is a double helix of two complementary strands formed by four nucleotides (bases): Adenine, Cytosine, Guanine and Thymine 2 of 31 Gene
PROTEIN MULTIPLE ALIGNMENT MOTIVATION: BACKGROUND: Marina Sirota
 Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
Marina Sirota MOTIVATION: PROTEIN MULTIPLE ALIGNMENT To study evolution on the genetic level across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein
A Design of a Hybrid System for DNA Sequence Alignment
 IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
IMECS 2008, 9-2 March, 2008, Hong Kong A Design of a Hybrid System for DNA Sequence Alignment Heba Khaled, Hossam M. Faheem, Tayseer Hasan, Saeed Ghoneimy Abstract This paper describes a parallel algorithm
Acceleration of the Smith-Waterman algorithm for DNA sequence alignment using an FPGA platform
 Acceleration of the Smith-Waterman algorithm for DNA sequence alignment using an FPGA platform Barry Strengholt Matthijs Brobbel Delft University of Technology Faculty of Electrical Engineering, Mathematics
Acceleration of the Smith-Waterman algorithm for DNA sequence alignment using an FPGA platform Barry Strengholt Matthijs Brobbel Delft University of Technology Faculty of Electrical Engineering, Mathematics
Pairwise Sequence Alignment. Zhongming Zhao, PhD
 Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Pairwise alignment II
 Pairwise alignment II Agenda - Previous Lesson: Minhala + Introduction - Review Dynamic Programming - Pariwise Alignment Biological Motivation Today: - Quick Review: Sequence Alignment (Global, Local,
Pairwise alignment II Agenda - Previous Lesson: Minhala + Introduction - Review Dynamic Programming - Pariwise Alignment Biological Motivation Today: - Quick Review: Sequence Alignment (Global, Local,
Gaps ATTACGTACTCCATG ATTACGT CATG. In an edit script we need 4 edit operations for the gap of length 4.
 Gaps ATTACGTACTCCATG ATTACGT CATG In an edit script we need 4 edit operations for the gap of length 4. In maximal score alignments we treat the dash " " like any other character, hence we charge the s(x,
Gaps ATTACGTACTCCATG ATTACGT CATG In an edit script we need 4 edit operations for the gap of length 4. In maximal score alignments we treat the dash " " like any other character, hence we charge the s(x,
BMI/CS 576 Fall 2015 Midterm Exam
 BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
BMI/CS 576 Fall 2015 Midterm Exam Prof. Colin Dewey Tuesday, October 27th, 2015 11:00am-12:15pm Name: KEY Write your answers on these pages and show your work. You may use the back sides of pages as necessary.
Sequencee Analysis Algorithms for Bioinformatics Applications
 Zagazig University Faculty of Engineering Computers and Systems Engineering Department Sequencee Analysis Algorithms for Bioinformatics Applications By Mohamed Al sayed Mohamed Ali Issa B.Sc in Computers
Zagazig University Faculty of Engineering Computers and Systems Engineering Department Sequencee Analysis Algorithms for Bioinformatics Applications By Mohamed Al sayed Mohamed Ali Issa B.Sc in Computers
A Revised Algorithm to find Longest Common Subsequence
 A Revised Algorithm to find Longest Common Subsequence Deena Nath 1, Jitendra Kurmi 2, Deveki Nandan Shukla 3 1, 2, 3 Department of Computer Science, Babasaheb Bhimrao Ambedkar University Lucknow Abstract:
A Revised Algorithm to find Longest Common Subsequence Deena Nath 1, Jitendra Kurmi 2, Deveki Nandan Shukla 3 1, 2, 3 Department of Computer Science, Babasaheb Bhimrao Ambedkar University Lucknow Abstract:
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT
 OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
OPEN MP-BASED PARALLEL AND SCALABLE GENETIC SEQUENCE ALIGNMENT Asif Ali Khan*, Laiq Hassan*, Salim Ullah* ABSTRACT: In bioinformatics, sequence alignment is a common and insistent task. Biologists align
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
1. R. Durbin, S. Eddy, A. Krogh und G. Mitchison: Biological sequence analysis, Cambridge, 1998
 7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
7 Multiple Sequence Alignment The exposition was prepared by Clemens GrÃP pl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
On the Efficacy of Haskell for High Performance Computational Biology
 On the Efficacy of Haskell for High Performance Computational Biology Jacqueline Addesa Academic Advisors: Jeremy Archuleta, Wu chun Feng 1. Problem and Motivation Biologists can leverage the power of
On the Efficacy of Haskell for High Performance Computational Biology Jacqueline Addesa Academic Advisors: Jeremy Archuleta, Wu chun Feng 1. Problem and Motivation Biologists can leverage the power of
Basics on bioinforma-cs Lecture 4. Concita Cantarella
 Basics on bioinforma-cs Lecture 4 Concita Cantarella concita.cantarella@entecra.it; concita.cantarella@gmail.com Why compare sequences Sequence comparison is a way of arranging the sequences of DNA, RNA
Basics on bioinforma-cs Lecture 4 Concita Cantarella concita.cantarella@entecra.it; concita.cantarella@gmail.com Why compare sequences Sequence comparison is a way of arranging the sequences of DNA, RNA
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
EECS730: Introduction to Bioinformatics Lecture 06: Multiple Sequence Alignment https://upload.wikimedia.org/wikipedia/commons/thumb/7/79/rplp0_90_clustalw_aln.gif/575px-rplp0_90_clustalw_aln.gif Slides
1. R. Durbin, S. Eddy, A. Krogh und G. Mitchison: Biological sequence analysis, Cambridge, 1998
 7 Multiple Sequence Alignment The exposition was prepared by Clemens Gröpl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
7 Multiple Sequence Alignment The exposition was prepared by Clemens Gröpl, based on earlier versions by Daniel Huson, Knut Reinert, and Gunnar Klau. It is based on the following sources, which are all
Sequence Alignment. Ulf Leser
 Sequence Alignment Ulf Leser his Lecture Approximate String Matching Edit distance and alignment Computing global alignments Local alignment Ulf Leser: Bioinformatics, Summer Semester 2016 2 ene Function
Sequence Alignment Ulf Leser his Lecture Approximate String Matching Edit distance and alignment Computing global alignments Local alignment Ulf Leser: Bioinformatics, Summer Semester 2016 2 ene Function
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS
 FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
FINDING APPROXIMATE REPEATS WITH MULTIPLE SPACED SEEDS FINDING APPROXIMATE REPEATS IN DNA SEQUENCES USING MULTIPLE SPACED SEEDS By SARAH BANYASSADY, B.S. A Thesis Submitted to the School of Graduate Studies
Sequence alignment algorithms
 Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
15.4 Longest common subsequence
 15.4 Longest common subsequence Biological applications often need to compare the DNA of two (or more) different organisms A strand of DNA consists of a string of molecules called bases, where the possible
15.4 Longest common subsequence Biological applications often need to compare the DNA of two (or more) different organisms A strand of DNA consists of a string of molecules called bases, where the possible
CS2220: Introduction to Computational Biology Lecture 5: Essence of Sequence Comparison. Limsoon Wong
 For written notes on this lecture, please read chapter 10 of The Practical Bioinformatician CS2220: Introduction to Computational Biology Lecture 5: Essence of Sequence Comparison Limsoon Wong 2 Plan Dynamic
For written notes on this lecture, please read chapter 10 of The Practical Bioinformatician CS2220: Introduction to Computational Biology Lecture 5: Essence of Sequence Comparison Limsoon Wong 2 Plan Dynamic
Lectures 12 and 13 Dynamic programming: weighted interval scheduling
 Lectures 12 and 13 Dynamic programming: weighted interval scheduling COMP 523: Advanced Algorithmic Techniques Lecturer: Dariusz Kowalski Lectures 12-13: Dynamic Programming 1 Overview Last week: Graph
Lectures 12 and 13 Dynamic programming: weighted interval scheduling COMP 523: Advanced Algorithmic Techniques Lecturer: Dariusz Kowalski Lectures 12-13: Dynamic Programming 1 Overview Last week: Graph
Distributed Protein Sequence Alignment
 Distributed Protein Sequence Alignment ABSTRACT J. Michael Meehan meehan@wwu.edu James Hearne hearne@wwu.edu Given the explosive growth of biological sequence databases and the computational complexity
Distributed Protein Sequence Alignment ABSTRACT J. Michael Meehan meehan@wwu.edu James Hearne hearne@wwu.edu Given the explosive growth of biological sequence databases and the computational complexity
Sequence Alignment: Mo1va1on and Algorithms. Lecture 2: August 23, 2012
 Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Cache and Energy Efficient Alignment of Very Long Sequences
 Cache and Energy Efficient Alignment of Very Long Sequences Chunchun Zhao Department of Computer and Information Science and Engineering University of Florida Email: czhao@cise.ufl.edu Sartaj Sahni Department
Cache and Energy Efficient Alignment of Very Long Sequences Chunchun Zhao Department of Computer and Information Science and Engineering University of Florida Email: czhao@cise.ufl.edu Sartaj Sahni Department
BGGN 213 Foundations of Bioinformatics Barry Grant
 BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Dynamic Programming II
 June 9, 214 DP: Longest common subsequence biologists often need to find out how similar are 2 DNA sequences DNA sequences are strings of bases: A, C, T and G how to define similarity? DP: Longest common
June 9, 214 DP: Longest common subsequence biologists often need to find out how similar are 2 DNA sequences DNA sequences are strings of bases: A, C, T and G how to define similarity? DP: Longest common
CSE 417 Dynamic Programming (pt 5) Multiple Inputs
 Multiple Inputs") CSE 417 Dynamic Programming (pt 5) Multiple Inputs Reminders > HW5 due Wednesday Dynamic Programming Review > Apply the steps... optimal substructure: (small) set of solutions, constructed from solutions
CSE 417 Dynamic Programming (pt 5) Multiple Inputs Reminders > HW5 due Wednesday Dynamic Programming Review > Apply the steps... optimal substructure: (small) set of solutions, constructed from solutions
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships
 Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
Comparison of Sequence Similarity Measures for Distant Evolutionary Relationships Abhishek Majumdar, Peter Z. Revesz Department of Computer Science and Engineering, University of Nebraska-Lincoln, Lincoln,
A multiple alignment tool in 3D
 Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
Outline Department of Computer Science, Bioinformatics Group University of Leipzig TBI Winterseminar Bled, Slovenia February 2005 Outline Outline 1 Multiple Alignments Problems Goal Outline Outline 1 Multiple
Alignment Based Similarity distance Measure for Better Web Sessions Clustering
 Available online at www.sciencedirect.com Procedia Computer Science 5 (2011) 450 457 The 2 nd International Conference on Ambient Systems, Networks and Technologies (ANT) Alignment Based Similarity distance
Available online at www.sciencedirect.com Procedia Computer Science 5 (2011) 450 457 The 2 nd International Conference on Ambient Systems, Networks and Technologies (ANT) Alignment Based Similarity distance
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Central Issues in Biological Sequence Comparison
 Central Issues in Biological Sequence Comparison Definitions: What is one trying to find or optimize? Algorithms: Can one find the proposed object optimally or in reasonable time optimize? Statistics:
Central Issues in Biological Sequence Comparison Definitions: What is one trying to find or optimize? Algorithms: Can one find the proposed object optimally or in reasonable time optimize? Statistics:
Multiple Sequence Alignment. With thanks to Eric Stone and Steffen Heber, North Carolina State University
 Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
Multiple Sequence Alignment With thanks to Eric Stone and Steffen Heber, North Carolina State University Definition: Multiple sequence alignment Given a set of sequences, a multiple sequence alignment
Algorithm Design and Analysis
 Algorithm Design and Analysis LECTURE 16 Dynamic Programming Least Common Subsequence Saving space Adam Smith Least Common Subsequence A.k.a. sequence alignment edit distance Longest Common Subsequence
Algorithm Design and Analysis LECTURE 16 Dynamic Programming Least Common Subsequence Saving space Adam Smith Least Common Subsequence A.k.a. sequence alignment edit distance Longest Common Subsequence
Multiple Sequence Alignment Augmented by Expert User Constraints
 Multiple Sequence Alignment Augmented by Expert User Constraints A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of
Multiple Sequence Alignment Augmented by Expert User Constraints A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database