Sequence Alignment: Mo1va1on and Algorithms
|
|
|
- Hortense McDowell
- 5 years ago
- Views:
Transcription
1 Sequence Alignment: Mo1va1on and Algorithms
2 Mo1va1on and Introduc1on
3 Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually implies significant func1onal or structural similarity. imply structure from the sequence similarity suggest gene func1on Frequently, we aren t interested in exact matches of the sequences but approximate matches.
4 Inexact Alignment Where is GATTACA approximately in the human genome? And how do we efficiently find them? Answers to these ques1ons depend on: 1. The defini1on of approximately Hamming Distance, Edit distance, Sequence Similarity 2. Efficiency depends on the data characteris1cs and goals
5 In this lecture Sequence alignment and alignment sotware are used all over bioinforma1cs for different purposes. The goal of this lecture is to understand sequence alignment, mo1vate it in the context of assembly, and seeing what sotware is out there.
6 Two examples where you would use sequence similarity 1. Use a known genome to help assemble an unknown genome. 2. Annota1ng a genome.
7 Two examples where you would use sequence similarity 1. Use a known genome to help assemble an unknown genome. 2. Annota1ng a genome.
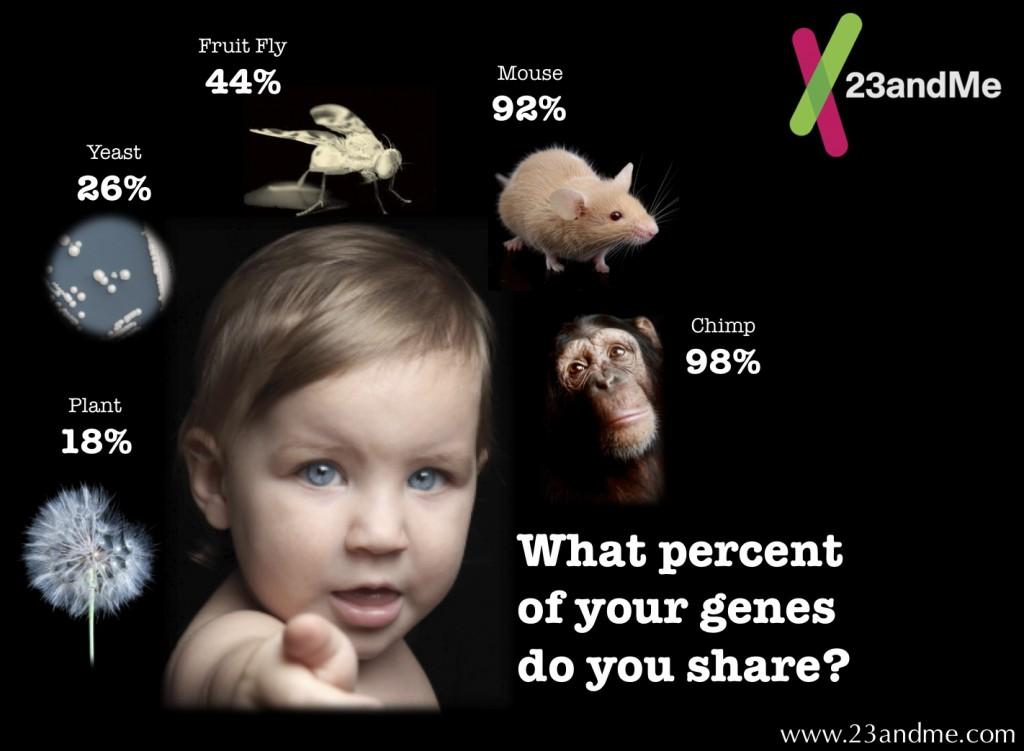
8 What is a reference genome? A reference genome is a representa1ve example of a species set of genes. As they are oten assembled from the sequencing of DNA from a number of donors, reference genomes do not accurately represent the set of genes of any individual. BUT the reference is very, very, VERY similar in many loca1ons to the individual.


9
10 Sample Prepara*on 10
11 Sample Prepara*on Fragments 11
 ACGTAGAATCGACCATG ACGTAGAATACGTAGAA GGGACGTAGAATACGAC Reads")
12 Sample Prepara*on Fragments Sequencing Next Generation Sequencing (NGS) ACGTAGAATCGACCATG ACGTAGAATACGTAGAA GGGACGTAGAATACGAC Reads 12
13 Sample Prepara*on Fragments Sequencing Reads Assembly ACGTAGAATACGTAGAA ACGTAGAATCGACCATG GGGACGTAGAATACGAC ACGTAGAATACGTAGAAACAGATTAGAGAG Contigs 13

14 Sample Prepara*on Fragments Sequencing Reads Assembly ACGTAGAATACGTAGAA ACGTAGAATCGACCATG GGGACGTAGAATACGAC BUT WE DON T HAVE TO BEGIN FROM SCRATCH!!! ACGTAGAATACGTAGAAACAGATTAGAGAG Contigs 14
15 How do we use the reference? The reads from the individual will be very similar but not iden1cal to the reference genome. Ideally, we would like to use the informa1on from the reference! Otherwise, we are throwing up valuable informa1on. Hence, the first step is to find where in the genome the sequences are similar to.
16 Two examples where you would use sequence similarity 1. Use a known genome to help assemble an unknown genome. 2. Annota9ng a genome.
17
18 Sequence Alignment Before we can make compara1ve statements about two sequences, we have to produce a pairwise sequence alignment What is the op1mal alignment between two sequences? Match/mismatch? Gaps/extensions? Is an op1mal alignment always significant?
19 Edit Distance Levenshtein (1966) introduced edit distance between two strings as the minimum number of elementary operations (insertions, deletions, and substitutions) to transform one string into the other d(v, w): the minimum number of operations required to transform v into w.
20 Edit Distance vs Hamming Distance Hamming distance always compares the i th le^er of v with the j th le^er of w V = ATATATAT W = TATATATA Edit distance may compare the i th le^er of v with the j th le^er of w V = -ATATATAT W = TATATATA- d(v, w)=8 computing Hamming distance is a trivial task d(v, w)=2 computing edit distance is a non-trivial task
21 Edit Distance vs Hamming Distance Hamming distance always compares the i th le^er of v with the j th le^er of w V = ATATATAT W = TATATATA Edit distance may compare the i th le^er of v with the j th le^er of w V = -ATATATAT W = TATATATA- d(v, w)=8 d(v, w)=2 (one insertion and one deletion) How to find what j goes with what i such that the score of the alignment is op1mized?
22 Edit Distance: Example TGCATAT! ATCCGAT in 5 steps TGCATAT! (delete last T) TGCATA! (delete last A) TGCAT! (insert A at front) ATGCAT! (subs1tute C for 3 rd G) ATCCAT! (insert G before last A) ATCCGAT (Done)
23 Compu1ng Edit Distance
24 Dynamic Programming Algorithm Dynamic Programming is a method for solving complex problems by breaking them down into simpler subproblems. Provides the best (op1mal) alignment between two sequences. Includes matches, mismatches, and gaps to maximize the number of matched characters. Score: match, mismatch, gap (affine vs. non- affine)
25 Dynamic Programming Visualized A T _ G T T A T _ A T C G T _ A _ C Corresponding path (0,0), (1,1), (2,2), (2,3), (3,4), (4,5), (5,5), (6,6), (7,6), (7,7)
26 Dynamic Programming Visualized and represent indels in v and w with score 0. represent matches with score 1. The score of the alignment path is 5.
27 Dynamic Programming Visualized Every path in the edit graph corresponds to an alignment:
28 Score for match: +1 Score for mismatch: 0 Score for gap: 0 Define Rules Let s i,j be the total score at posi1on i, j then we have that it is equal to the maximum of {s i- 1, j (if v i = w j ), s i- 1, j (otherwise: gap or mismatch)}
29 Dynamic Programming Example Ini1alize 1 st row and 1 st column to be all zeroes. Or, to be more precise, ini1alize 0 th row and 0 th column to be all zeroes.
30 Dynamic Programming Example S i,j = maximum of : S i-1, j-1 S i-1, j S i, j-1 "value from NW +1, if v i = w j " value from North (top) " value from West (left)
31 Alignment: Backtracking Arrows show where the score originated. if from the top if from the let if v i = w j
32 Backtracking Example Find a match in row and column 2. i=2, j=2,5 is a match (T). j=2, i=4,5,7 is a match (T). Since v i = w j, s i,j = s i-1,j-1 +1 s 2,2 = [s 1,1 = 1] + 1 s 2,5 = [s 1,4 = 1] + 1 s 4,2 = [s 3,1 = 1] + 1 s 5,2 = [s 4,1 = 1] + 1 s 7,2 = [s 6,1 = 1] + 1
33 Backtracking Example Con1nuing with the dynamic programming algorithm gives this result.
34 Scoring Matrices The alignment score represent odds of obtaining that score between sequences known to be related to that obtained by chance alignment between unrelated sequences When the correct scoring matrix is used, alignment sta1s1cs are meaningful Different scoring schemes for DNA and protein sequences
35 Examples of Scoring Matrices Amino acid subs1tu1on matrices: PAM BLOSUM DNA subs1tu1on matrices: DNA is less conserved than protein sequences. Less effec1ve to compare coding regions at nucleo1de level.
36 Scoring Indels: Naive Approach A fixed penalty σ is given to every indel: - σ for 1 indel, - 2σ for 2 consecu1ve indels - 3σ for 3 consecu1ve indels, etc. Can be too severe penalty for a series of 100 consecu1ve indels
37 Affine Gap Penal1es A series of k indels oten come as a single event rather than a series of k single nucleo1de events: This is more likely. Normal scoring would give the same score for both alignments This is less likely.
38 Accoun1ng for Gaps Score for a gap of length x is - (ρ + σx), where ρ >0 is the penalty for introducing a gap: gap opening penalty ρ will be large rela1ve to σ: gap extension penalty because you do not want to add too much of a penalty for extending the gap.
39 Affine Gap Penal1es Gap penal1es: - ρ- σ when there is 1 indel - ρ- 2σ when there are 2 indels - ρ- 3σ when there are 3 indels, etc. - ρ- x σ (- gap opening - x gap extensions) Somehow reduced penal1es (as compared to naïve scoring) are given to runs of horizontal and ver1cal edges
40 Heuris1c Algorithms Make reasonable assump1ons about nature of sequence alignments and try out only most likely Make alignments reasonable assump1ons about nature of sequence alignments and try out only most likely alignments Full perfect match (word, k- tuple) Much faster than DP algorithms but less sensi1ve Extend alignment un1l:
41 Exis1ng Tools
42 Different Sequence Alignment Database Search: BLAST, FASTA, HMMER Mul1ple Sequence Alignment: ClustalW, FSA Genomic Analysis: BLAT Short Read Sequence Alignment: BWA, Bow9e, drfast, GSNAP, SHRiMP, SOAP, MAQ
43 BLAST Basic Local Alignment Search Tool BLAST is faster than Smith- Waterman (DP algorithm), it cannot guarantee the op1mal alignments of the query and database sequences. A BLAST search enables a researcher to compare a query sequence with a library or database of sequences and iden1fy library sequences that resemble the query sequence above a certain threshold. Different types of BLASTs are available according to the query sequences.
44 Blast Versions BLASTN: Compares a nucleo1de query to a nucleo1de database BLASTP: Compares a protein query to a protein database BLASTX: Compares a translated nucleo1de query to protein database TBLASTN: Compares a protein query to a translated nucleo1de database TBLASTX: Compares a translated nucleo1de query to a translated nucleo1de database

45 hip://blast.ncbi.nlm.nih.gov/
46 Typical Uses of BLAST Which bacterial species have a protein that is related in lineage to a certain protein with known amino- acid sequence? Where does a certain sequence of DNA originate? What other genes encode proteins that exhibit structures or mo1fs such as ones thtat have been determined?
47 Mul1ple Alignment A mul1ple sequence alignment is a sequence alignment of three or more biological sequences (protein, DNA, RNA) The query sequences are assumed to have some sort of evolu1onary rela1onship by which they share some sort of lineage Many tools are available but this is a really complex computa1onal problem Used in phylogene1cs
48 Short Read Alignment SW Bow9e: memory- efficient short read aligner. It aligns short DNA sequences (reads) to the human genome at a rate of over 25 million 35- bp reads per hours Burrows- Wheeler Aligner (BWA): an aliger that implements two algorithms: bwa- short and BWA- SW. The former works for query sequences shorter than 200bp and the la^er for longer sequences up to around 100kbp.



49 Sequence Alignment/Map Format Sequence Reads Alignment SoMware SAM File Resequencing RNA Seq SNPs
50 SAM format A tab- delimited text format consis1ng of a header sec1on, which is op1onal, and an alignment sec1on Example VN:1.0 SN:1 LN: AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta SN:2 LN: AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta SN:3 LN: AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta ID:UM0098:1 PL:ILLUMINA PU:HWUSI-EAS LHAAXX-L001 LB:80 DT: T20:00: SM:SD37 Example Alignments: 1:497:R: M17D24M M CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 19:20389:F:275+18M2D19M M = TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9 RG:Z:UM0098:1 XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 19:20389:F:275+18M2D19M M2D19M = GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:18^CA19 9: M2I25M:R: M2I27M = CACCACATCACATATACCAAGCCTGGCTGTGTCTTCT <;9<<5><<<<><<<>><<><>><9>><>>>9>>><> XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:5 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:35
51 The Alignment Column
52 Bitwise Flags FLAG: bitwise FLAG. Each bit is explained in the following table:
53 Bitwise Representa1on 1 = ! paired- end read 2 = ! mapped as proper pair 4 = ! unmapped read 8 = ! read mate unmapped 16 = ! read mapped on reverse strand Example: The flag 11! = (condi1ons 1, 2, 8) Flags 0, 4, and 16 are the flags most commonly used.
54 Mapping Quality Phred score, iden1cal to the quality measure in the fastq file. quality Q, probability P: P = 10 ^ (- Q / 10.0) If Q=30, P=1/1000!on average, one of out 1000 alignments will be wrong As good as this sounds it is not easy to compute such a quality.
55 Mapping Quality The repeat structure of the reference. Reads falling in repe11ve regions usually get very low mapping quality The base quality of the read. Low quality means the observed read sequence is possibly wrong, and wrong sequence may lead to a wrong alignment The sensi1vity of the alignment algorithm. The true hit is more likely to be missed by an algorithm with low sensi1vity, which also causes mapping errors Paired end or not. Reads mapped in pairs are more likely to be correct
56 BWA Specific High Scores A read alignment with a mapping quality 30 or above usually implies: The overall base quality of the read is good. The best alignment has few mismatches. The read has few or just one good hit on the reference, which means the current alignment is s1ll the best even if one or two bases are actually muta1ons or sequencing errors.
57 BWA Specific Low Scores Surprisingly difficult to track down the exact behavior Q=0!if a read can be aligned equally well to mul1ple posi1ons, BWA will randomly pick one posi1on and give it a mapping quality zero. Q=25!the edit distance equals mismatches and is greater than zero
58 What to do with low quality scores? Find repeat structures in the genome/con1g. Determine if there is a problem with your alignment or data (i.e. all the reads mapped with low quality scores). Filter them out. Very common to write a perl/ python script to filter out poorly aligned reads. Many, many, many other possibili1es.


59 The Alignment Column
60 CIGAR String CIGAR string is a compact representa1on of how the read aligned to the reference genome at that exact posi1on. More specifically, the CIGAR string is a sequence of of base lengths and the associated opera1on. match/mismatch with the reference. deleted/inserted from the from the reference.
61 Example of CIGAR RefPos: Reference: C C A T A C T G A A C T G A C T A A C Read: A C T A G A A T G G C T In the SAM file you will have the following fields: POS: 5 CIGAR: 3M1I3M1D5M
62 Final Comments BAM is a compressed version of the SAM file format. There are mul1ple programs that convert BAM files to SAM files and vice versa. Tablet (h^p://bioinf.scri.ac.uk/tablet/) is an easy to use, program that allows you to visualize an alignment. You simply give it a sam file and a fasta file and it reads the sam file and shows you the alignment.
Sequence Alignment: Mo1va1on and Algorithms. Lecture 2: August 23, 2012
 Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
Sequence Alignment: Mo1va1on and Algorithms Lecture 2: August 23, 2012 Mo1va1on and Introduc1on Importance of Sequence Alignment For DNA, RNA and amino acid sequences, high sequence similarity usually
EECS 4425: Introductory Computational Bioinformatics Fall Suprakash Datta
 EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
EECS 4425: Introductory Computational Bioinformatics Fall 2018 Suprakash Datta datta [at] cse.yorku.ca Office: CSEB 3043 Phone: 416-736-2100 ext 77875 Course page: http://www.cse.yorku.ca/course/4425 Many
Read Mapping and Variant Calling
 Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
Read Mapping and Variant Calling Whole Genome Resequencing Sequencing mul:ple individuals from the same species Reference genome is already available Discover varia:ons in the genomes between and within
Mar%n Norling. Uppsala, November 15th 2016
 Mar%n Norling Uppsala, November 15th 2016 What can we do with an assembly? Since we can never know the actual sequence, or its varia%ons, valida%ng an assembly is tricky. But once you ve used all the assemblers,
Mar%n Norling Uppsala, November 15th 2016 What can we do with an assembly? Since we can never know the actual sequence, or its varia%ons, valida%ng an assembly is tricky. But once you ve used all the assemblers,
Module: Sequence Alignment Theory and Applica8ons Session: BLAST
 Module: Sequence Alignment Theory and Applica8ons Session: BLAST Learning Objec8ves and Outcomes v Understand the principles of the BLAST algorithm v Understand the different BLAST algorithms, parameters
Module: Sequence Alignment Theory and Applica8ons Session: BLAST Learning Objec8ves and Outcomes v Understand the principles of the BLAST algorithm v Understand the different BLAST algorithms, parameters
NGS Data and Sequence Alignment
 Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
Applications and Servers SERVER/REMOTE Compute DB WEB Data files NGS Data and Sequence Alignment SSH WEB SCP Manpreet S. Katari App Aug 11, 2016 Service Terminal IGV Data files Window Personal Computer/Local
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, This lecture is based on the following papers, which are all recommended reading:
 24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
24 Grundlagen der Bioinformatik, SS 10, D. Huson, April 26, 2010 3 BLAST and FASTA This lecture is based on the following papers, which are all recommended reading: D.J. Lipman and W.R. Pearson, Rapid
Basic Local Alignment Search Tool (BLAST)
") BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST 26.04.2018 Basic Local Alignment Search Tool (BLAST) BLAST (Altshul-1990) is an heuristic Pairwise Alignment composed by six-steps that search for local similarities. The most used access point to
BLAST MCDB 187. Friday, February 8, 13
 BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
BLAST MCDB 187 BLAST Basic Local Alignment Sequence Tool Uses shortcut to compute alignments of a sequence against a database very quickly Typically takes about a minute to align a sequence against a database
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines 454 GS Junior,
File Formats: SAM, BAM, and CRAM. UCD Genome Center Bioinformatics Core Tuesday 15 September 2015
 File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
File Formats: SAM, BAM, and CRAM UCD Genome Center Bioinformatics Core Tuesday 15 September 2015 / BAM / CRAM NEW! http://samtools.sourceforge.net/ - deprecated! http://www.htslib.org/ - SAMtools 1.0 and
Lecture 5 Advanced BLAST
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 5 Advanced BLAST BLAST Recap Sequence Alignment Complexity and indexing BLASTN and BLASTP Basic parameters
High-throughput sequencing: Alignment and related topic. Simon Anders EMBL Heidelberg
 High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
High-throughput sequencing: Alignment and related topic Simon Anders EMBL Heidelberg Established platforms HTS Platforms Illumina HiSeq, ABI SOLiD, Roche 454 Newcomers: Benchtop machines: Illumina MiSeq,
As of August 15, 2008, GenBank contained bases from reported sequences. The search procedure should be
 48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
48 Bioinformatics I, WS 09-10, S. Henz (script by D. Huson) November 26, 2009 4 BLAST and BLAT Outline of the chapter: 1. Heuristics for the pairwise local alignment of two sequences 2. BLAST: search and
Biology 644: Bioinformatics
 Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Find the best alignment between 2 sequences with lengths n and m, respectively Best alignment is very dependent upon the substitution matrix and gap penalties The Global Alignment Problem tries to find
Lecture 12. Short read aligners
 Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
Lecture 12 Short read aligners Ebola reference genome We will align ebola sequencing data against the 1976 Mayinga reference genome. We will hold the reference gnome and all indices: mkdir -p ~/reference/ebola
The SAM Format Specification (v1.3 draft)
") The SAM Format Specification (v1.3 draft) The SAM Format Specification Working Group July 15, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
The SAM Format Specification (v1.3 draft) The SAM Format Specification Working Group July 15, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited text
SAM : Sequence Alignment/Map format. A TAB-delimited text format storing the alignment information. A header section is optional.
 Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Alignment of NGS reads, samtools and visualization Hands-on Software used in this practical BWA MEM : Burrows-Wheeler Aligner. A software package for mapping low-divergent sequences against a large reference
Sequence mapping and assembly. Alistair Ward - Boston College
 Sequence mapping and assembly Alistair Ward - Boston College Sequenced a genome? Fragmented a genome -> DNA library PCR amplification Sequence reads (ends of DNA fragment for mate pairs) We no longer have
Sequence mapping and assembly Alistair Ward - Boston College Sequenced a genome? Fragmented a genome -> DNA library PCR amplification Sequence reads (ends of DNA fragment for mate pairs) We no longer have
Database Similarity Searching
 Database Similarity Searching Why search databases? To find out if a new DNA sequence shares similari?es with sequences already deposited in the databanks. To find proteins homologous to a puta?ve coding
Database Similarity Searching Why search databases? To find out if a new DNA sequence shares similari?es with sequences already deposited in the databanks. To find proteins homologous to a puta?ve coding
Basics on bioinforma-cs Lecture 4. Concita Cantarella
 Basics on bioinforma-cs Lecture 4 Concita Cantarella concita.cantarella@entecra.it; concita.cantarella@gmail.com Why compare sequences Sequence comparison is a way of arranging the sequences of DNA, RNA
Basics on bioinforma-cs Lecture 4 Concita Cantarella concita.cantarella@entecra.it; concita.cantarella@gmail.com Why compare sequences Sequence comparison is a way of arranging the sequences of DNA, RNA
Finding sequence similarities with genes of known function is a common approach to infer a newly sequenced gene s function
 Outline DNA Sequence Comparison: First Success Stories Change Problem Manhattan Tourist Problem Longest Paths in Graphs Sequence Alignment Edit Distance Longest Common Subsequence Problem Dot Matrices
Outline DNA Sequence Comparison: First Success Stories Change Problem Manhattan Tourist Problem Longest Paths in Graphs Sequence Alignment Edit Distance Longest Common Subsequence Problem Dot Matrices
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
EECS730: Introduction to Bioinformatics Lecture 04: Variations of sequence alignments http://www.pitt.edu/~mcs2/teaching/biocomp/tutorials/global.html Slides adapted from Dr. Shaojie Zhang (University
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies May 15, 2014 1 BLAST What is BLAST? The algorithm 2 Genome assembly De novo assembly Mapping assembly 3
INTRODUCTION AUX FORMATS DE FICHIERS
 INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
INTRODUCTION AUX FORMATS DE FICHIERS Plan. Formats de séquences brutes.. Format fasta.2. Format fastq 2. Formats d alignements 2.. Format SAM 2.2. Format BAM 4. Format «Variant Calling» 4.. Format Varscan
BLAST & Genome assembly
 BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
BLAST & Genome assembly Solon P. Pissis Tomáš Flouri Heidelberg Institute for Theoretical Studies November 17, 2012 1 Introduction Introduction 2 BLAST What is BLAST? The algorithm 3 Genome assembly De
Bioinformatics. Sequence alignment BLAST Significance. Next time Protein Structure
 Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
Bioinformatics Sequence alignment BLAST Significance Next time Protein Structure 1 Experimental origins of sequence data The Sanger dideoxynucleotide method F Each color is one lane of an electrophoresis
BLAST, Profile, and PSI-BLAST
 BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
BLAST, Profile, and PSI-BLAST Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 26 Free for academic use Copyright @ Jianlin Cheng & original sources
Database Searching Using BLAST
 Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Mahidol University Objectives SCMI512 Molecular Sequence Analysis Database Searching Using BLAST Lecture 2B After class, students should be able to: explain the FASTA algorithm for database searching explain
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Director Bioinformatics & Research Computing Whitehead Institute Topics to Cover
The SAM Format Specification (v1.3-r837)
") The SAM Format Specification (v1.3-r837) The SAM Format Specification Working Group November 18, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited
The SAM Format Specification (v1.3-r837) The SAM Format Specification Working Group November 18, 2010 1 The SAM Format Specification SAM stands for Sequence Alignment/Map format. It is a TAB-delimited
Short Read Alignment. Mapping Reads to a Reference
 Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
Short Read Alignment Mapping Reads to a Reference Brandi Cantarel, Ph.D. & Daehwan Kim, Ph.D. BICF 05/2018 Introduction to Mapping Short Read Aligners DNA vs RNA Alignment Quality Pitfalls and Improvements
NGS Data Analysis. Roberto Preste
 NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
NGS Data Analysis Roberto Preste 1 Useful info http://bit.ly/2r1y2dr Contacts: roberto.preste@gmail.com Slides: http://bit.ly/ngs-data 2 NGS data analysis Overview 3 NGS Data Analysis: the basic idea http://bit.ly/2r1y2dr
Introduction to Computational Molecular Biology
 18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
18.417 Introduction to Computational Molecular Biology Lecture 13: October 21, 2004 Scribe: Eitan Reich Lecturer: Ross Lippert Editor: Peter Lee 13.1 Introduction We have been looking at algorithms to
High-throughout sequencing and using short-read aligners. Simon Anders
 High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
High-throughout sequencing and using short-read aligners Simon Anders High-throughput sequencing (HTS) Sequencing millions of short DNA fragments in parallel. a.k.a.: next-generation sequencing (NGS) massively-parallel
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
Computational Genomics and Molecular Biology, Fall 2015 1 Sequence Alignment Dannie Durand Pairwise Sequence Alignment The goal of pairwise sequence alignment is to establish a correspondence between the
RNA-seq. Manpreet S. Katari
 RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
RNA-seq Manpreet S. Katari Evolution of Sequence Technology Normalizing the Data RPKM (Reads per Kilobase of exons per million reads) Score = R NT R = # of unique reads for the gene N = Size of the gene
Bioinformatics explained: BLAST. March 8, 2007
 Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Bioinformatics Explained Bioinformatics explained: BLAST March 8, 2007 CLC bio Gustav Wieds Vej 10 8000 Aarhus C Denmark Telephone: +45 70 22 55 09 Fax: +45 70 22 55 19 www.clcbio.com info@clcbio.com Bioinformatics
Pairwise Sequence Alignment. Zhongming Zhao, PhD
 Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Pairwise Sequence Alignment Zhongming Zhao, PhD Email: zhongming.zhao@vanderbilt.edu http://bioinfo.mc.vanderbilt.edu/ Sequence Similarity match mismatch A T T A C G C G T A C C A T A T T A T G C G A T
Mapping NGS reads for genomics studies
 Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Mapping NGS reads for genomics studies Valencia, 28-30 Sep 2015 BIER Alejandro Alemán aaleman@cipf.es Genomics Data Analysis CIBERER Where are we? Fastq Sequence preprocessing Fastq Alignment BAM Visualization
Sequence alignment theory and applications Session 3: BLAST algorithm
 Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence alignment theory and applications Session 3: BLAST algorithm Introduction to Bioinformatics online course : IBT Sonal Henson Learning Objectives Understand the principles of the BLAST algorithm
Sequence Alignment. GS , Introduc8on to Bioinforma8cs The University of Texas GSBS program, Fall 2013
 Sequence Alignment GS 011143, Introduc8on to Bioinforma8cs The University of Texas GSBS program, Fall 2013 Ken Chen, Ph.D. Department of Bioinforma8cs and Computa8onal Biology UT MD Anderson Cancer Center
Sequence Alignment GS 011143, Introduc8on to Bioinforma8cs The University of Texas GSBS program, Fall 2013 Ken Chen, Ph.D. Department of Bioinforma8cs and Computa8onal Biology UT MD Anderson Cancer Center
Sequencing. Short Read Alignment. Sequencing. Paired-End Sequencing 6/10/2010. Tobias Rausch 7 th June 2010 WGS. ChIP-Seq. Applied Biosystems.
 Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequencing Short Alignment Tobias Rausch 7 th June 2010 WGS RNA-Seq Exon Capture ChIP-Seq Sequencing Paired-End Sequencing Target genome Fragments Roche GS FLX Titanium Illumina Applied Biosystems SOLiD
Sequence Alignment. GBIO0002 Archana Bhardwaj University of Liege
 Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Sequence Alignment GBIO0002 Archana Bhardwaj University of Liege 1 What is Sequence Alignment? A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity.
Lecture Overview. Sequence search & alignment. Searching sequence databases. Sequence Alignment & Search. Goals: Motivations:
 Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Lecture Overview Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating
Wilson Leung 01/03/2018 An Introduction to NCBI BLAST. Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment
 An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
An Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at https://blast.ncbi.nlm.nih.gov/blast.cgi
Sequence Alignment & Search
 Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Sequence Alignment & Search Karin Verspoor, Ph.D. Faculty, Computational Bioscience Program University of Colorado School of Medicine With credit and thanks to Larry Hunter for creating the first version
Similarity Searches on Sequence Databases
 Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Similarity Searches on Sequence Databases Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Zürich, October 2004 Swiss Institute of Bioinformatics Swiss EMBnet node Outline Importance of
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Computational Molecular Biology Erwin M. Bakker Lecture 3, mainly from material by R. Shamir [2] and H.J. Hoogeboom [4]. 1 Pairwise Sequence Alignment Biological Motivation Algorithmic Aspect Recursive
Principles of Bioinformatics. BIO540/STA569/CSI660 Fall 2010
 Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Principles of Bioinformatics BIO540/STA569/CSI660 Fall 2010 Lecture 11 Multiple Sequence Alignment I Administrivia Administrivia The midterm examination will be Monday, October 18 th, in class. Closed
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA.
 Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence or library of DNA. Fasta is used to compare a protein or DNA sequence to all of the
Under the Hood of Alignment Algorithms for NGS Researchers
 Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
Under the Hood of Alignment Algorithms for NGS Researchers April 16, 2014 Gabe Rudy VP of Product Development Golden Helix Questions during the presentation Use the Questions pane in your GoToWebinar window
BGGN 213 Foundations of Bioinformatics Barry Grant
 BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
BGGN 213 Foundations of Bioinformatics Barry Grant http://thegrantlab.org/bggn213 Recap From Last Time: 25 Responses: https://tinyurl.com/bggn213-02-f17 Why ALIGNMENT FOUNDATIONS Why compare biological
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD
 Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
Lecture 2 Pairwise sequence alignment. Principles Computational Biology Teresa Przytycka, PhD Assumptions: Biological sequences evolved by evolution. Micro scale changes: For short sequences (e.g. one
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar..
 .. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
.. Fall 2011 CSC 570: Bioinformatics Alexander Dekhtyar.. PAM and BLOSUM Matrices Prepared by: Jason Banich and Chris Hoover Background As DNA sequences change and evolve, certain amino acids are more
Next generation sequencing: assembly by mapping reads. Laurent Falquet, Vital-IT Helsinki, June 3, 2010
 Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Next generation sequencing: assembly by mapping reads Laurent Falquet, Vital-IT Helsinki, June 3, 2010 Overview What is assembly by mapping? Methods BWT File formats Tools Issues Visualization Discussion
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014
 Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
Dynamic Programming User Manual v1.0 Anton E. Weisstein, Truman State University Aug. 19, 2014 Dynamic programming is a group of mathematical methods used to sequentially split a complicated problem into
B L A S T! BLAST: Basic local alignment search tool. Copyright notice. February 6, Pairwise alignment: key points. Outline of tonight s lecture
 February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
February 6, 2008 BLAST: Basic local alignment search tool B L A S T! Jonathan Pevsner, Ph.D. Introduction to Bioinformatics pevsner@jhmi.edu 4.633.0 Copyright notice Many of the images in this powerpoint
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties
 Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
Global Alignment Scoring Matrices Local Alignment Alignment with Affine Gap Penalties From LCS to Alignment: Change the Scoring The Longest Common Subsequence (LCS) problem the simplest form of sequence
PPI Network Alignment Advanced Topics in Computa8onal Genomics
 PPI Network Alignment 02-715 Advanced Topics in Computa8onal Genomics PPI Network Alignment Compara8ve analysis of PPI networks across different species by aligning the PPI networks Find func8onal orthologs
PPI Network Alignment 02-715 Advanced Topics in Computa8onal Genomics PPI Network Alignment Compara8ve analysis of PPI networks across different species by aligning the PPI networks Find func8onal orthologs
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Sequence pairwise alignment Score statistics: E-value and p-value Heuristic algorithms: BLAST and FASTA Database search: gene finding and annotations
FASTA. Besides that, FASTA package provides SSEARCH, an implementation of the optimal Smith- Waterman algorithm.
 FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
FASTA INTRODUCTION Definition (by David J. Lipman and William R. Pearson in 1985) - Compares a sequence of protein to another sequence or database of a protein, or a sequence of DNA to another sequence
TCCAGGTG-GAT TGCAAGTGCG-T. Local Sequence Alignment & Heuristic Local Aligners. Review: Probabilistic Interpretation. Chance or true homology?
 Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Local Sequence Alignment & Heuristic Local Aligners Lectures 18 Nov 28, 2011 CSE 527 Computational Biology, Fall 2011 Instructor: Su-In Lee TA: Christopher Miles Monday & Wednesday 12:00-1:20 Johnson Hall
Perl for Biologists. Session 8. April 30, Practical examples. (/home/jarekp/perl_08) Jon Zhang
 Jon Zhang") Perl for Biologists Session 8 April 30, 2014 Practical examples (/home/jarekp/perl_08) Jon Zhang Session 8: Examples CBSU Perl for Biologists 1.1 1 Review of Session 7 Regular expression: a specific pattern
Perl for Biologists Session 8 April 30, 2014 Practical examples (/home/jarekp/perl_08) Jon Zhang Session 8: Examples CBSU Perl for Biologists 1.1 1 Review of Session 7 Regular expression: a specific pattern
Alignments BLAST, BLAT
 Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
Alignments BLAST, BLAT Genome Genome Gene vs Built of DNA DNA Describes Organism Protein gene Stored as Circular/ linear Single molecule, or a few of them Both (depending on the species) Part of genome
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2011 Once sequenced the problem becomes computational
RCAC. Job files Example: Running seqyclean (a module)
") RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
RCAC Job files Why? When you log into an RCAC server you are using a special server designed for multiple users. This is called a frontend node ( or sometimes a head node). There are (I think) three front
Heuristic methods for pairwise alignment:
 Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Bi03c_1 Unit 03c: Heuristic methods for pairwise alignment: k-tuple-methods k-tuple-methods for alignment of pairs of sequences Bi03c_2 dynamic programming is too slow for large databases Use heuristic
Wilson Leung 05/27/2008 A Simple Introduction to NCBI BLAST
 A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
A Simple Introduction to NCBI BLAST Prerequisites: Detecting and Interpreting Genetic Homology: Lecture Notes on Alignment Resources: The BLAST web server is available at http://www.ncbi.nih.gov/blast/
NGS Analysis Using Galaxy
 NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
NGS Analysis Using Galaxy Sequences and Alignment Format Galaxy overview and Interface Get;ng Data in Galaxy Analyzing Data in Galaxy Quality Control Mapping Data History and workflow Galaxy Exercises
From Smith-Waterman to BLAST
 From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
From Smith-Waterman to BLAST Jeremy Buhler July 23, 2015 Smith-Waterman is the fundamental tool that we use to decide how similar two sequences are. Isn t that all that BLAST does? In principle, it is
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores.
 CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
CS 284A: Algorithms for Computational Biology Notes on Lecture: BLAST. The statistics of alignment scores. prepared by Oleksii Kuchaiev, based on presentation by Xiaohui Xie on February 20th. 1 Introduction
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2019 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Computational Molecular Biology
 Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Computational Molecular Biology Erwin M. Bakker Lecture 2 Materials used from R. Shamir [2] and H.J. Hoogeboom [4]. 1 Molecular Biology Sequences DNA A, T, C, G RNA A, U, C, G Protein A, R, D, N, C E,
Genomic Files. University of Massachusetts Medical School. October, 2014
 .. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2014 2 / 39. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
Genomic Files. University of Massachusetts Medical School. October, 2015
 .. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
.. Genomic Files University of Massachusetts Medical School October, 2015 2 / 55. A Typical Deep-Sequencing Workflow Samples Fastq Files Fastq Files Sam / Bam Files Various files Deep Sequencing Further
INTRODUCTION TO BIOINFORMATICS
 Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Molecular Biology-2017 1 INTRODUCTION TO BIOINFORMATICS In this section, we want to provide a simple introduction to using the web site of the National Center for Biotechnology Information NCBI) to obtain
Chapter 4: Blast. Chaochun Wei Fall 2014
 Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
Course organization Introduction ( Week 1-2) Course introduction A brief introduction to molecular biology A brief introduction to sequence comparison Part I: Algorithms for Sequence Analysis (Week 3-11)
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1. Database Searching
 COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
COS 551: Introduction to Computational Molecular Biology Lecture: Oct 17, 2000 Lecturer: Mona Singh Scribe: Jacob Brenner 1 Database Searching In database search, we typically have a large sequence database
RNA-Seq in Galaxy: Tuxedo protocol. Igor Makunin, UQ RCC, QCIF
 RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
RNA-Seq in Galaxy: Tuxedo protocol Igor Makunin, UQ RCC, QCIF Acknowledgments Genomics Virtual Lab: gvl.org.au Galaxy for tutorials: galaxy-tut.genome.edu.au Galaxy Australia: galaxy-aust.genome.edu.au
Today s Lecture. Multiple sequence alignment. Improved scoring of pairwise alignments. Affine gap penalties Profiles
 Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Today s Lecture Multiple sequence alignment Improved scoring of pairwise alignments Affine gap penalties Profiles 1 The Edit Graph for a Pair of Sequences G A C G T T G A A T G A C C C A C A T G A C G
Sequence alignment algorithms
 Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Sequence alignment algorithms Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 23 rd 27 After this lecture, you can decide when to use local and global sequence alignments
Illumina Next Generation Sequencing Data analysis
 Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
Illumina Next Generation Sequencing Data analysis Chiara Dal Fiume Sr Field Application Scientist Italy 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense Out of Life,
MetaPhyler Usage Manual
 MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
MetaPhyler Usage Manual Bo Liu boliu@umiacs.umd.edu March 13, 2012 Contents 1 What is MetaPhyler 1 2 Installation 1 3 Quick Start 2 3.1 Taxonomic profiling for metagenomic sequences.............. 2 3.2
Similarity searches in biological sequence databases
 Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Similarity searches in biological sequence databases Volker Flegel september 2004 Page 1 Outline Keyword search in databases General concept Examples SRS Entrez Expasy Similarity searches in databases
Read Mapping. Slides by Carl Kingsford
 Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
Read Mapping Slides by Carl Kingsford Bowtie Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Ben Langmead, Cole Trapnell, Mihai Pop and Steven L Salzberg, Genome Biology
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio CS 466 Saurabh Sinha
 BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
BLAST: Basic Local Alignment Search Tool Altschul et al. J. Mol Bio. 1990. CS 466 Saurabh Sinha Motivation Sequence homology to a known protein suggest function of newly sequenced protein Bioinformatics
Dynamic Programming & Smith-Waterman algorithm
 m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
m m Seminar: Classical Papers in Bioinformatics May 3rd, 2010 m m 1 2 3 m m Introduction m Definition is a method of solving problems by breaking them down into simpler steps problem need to contain overlapping
Reconstructing long sequences from overlapping sequence fragment. Searching databases for related sequences and subsequences
 SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
SEQUENCE ALIGNMENT ALGORITHMS 1 Why compare sequences? Reconstructing long sequences from overlapping sequence fragment Searching databases for related sequences and subsequences Storing, retrieving and
Algorithmic Approaches for Biological Data, Lecture #20
 Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Algorithmic Approaches for Biological Data, Lecture #20 Katherine St. John City University of New York American Museum of Natural History 20 April 2016 Outline Aligning with Gaps and Substitution Matrices
Introduction to Phylogenetics Week 2. Databases and Sequence Formats
 Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
Introduction to Phylogenetics Week 2 Databases and Sequence Formats I. Databases Crucial to bioinformatics The bigger the database, the more comparative research data Requires scientists to upload data
Introduc)on to annota)on with Artemis. Download presenta.on and data
on to annota)on with Artemis. Download presenta.on and data") Introduc)on to annota)on with Artemis Download presenta.on and data Annota)on Assign an informa)on to genomic sequences???? Genome annota)on 1. Iden.fying genomic elements by: Predic)on (structural annota.on
Introduc)on to annota)on with Artemis Download presenta.on and data Annota)on Assign an informa)on to genomic sequences???? Genome annota)on 1. Iden.fying genomic elements by: Predic)on (structural annota.on
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page.
 tutorial page.") Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Welcome to MAPHiTS (Mapping Analysis Pipeline for High-Throughput Sequences) tutorial page. In this page you will learn to use the tools of the MAPHiTS suite. A little advice before starting : rename your
Sequence Alignment (chapter 6) p The biological problem p Global alignment p Local alignment p Multiple alignment
 p The biological problem p Global alignment p Local alignment p Multiple alignment") Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
Sequence lignment (chapter 6) p The biological problem p lobal alignment p Local alignment p Multiple alignment Local alignment: rationale p Otherwise dissimilar proteins may have local regions of similarity
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection
 CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
CBSU/3CPG/CVG Joint Workshop Series Reference genome based sequence variation detection Computational Biology Service Unit (CBSU) Cornell Center for Comparative and Population Genomics (3CPG) Center for
Accurate Long-Read Alignment using Similarity Based Multiple Pattern Alignment and Prefix Tree Indexing
 Proposal for diploma thesis Accurate Long-Read Alignment using Similarity Based Multiple Pattern Alignment and Prefix Tree Indexing Astrid Rheinländer 01-09-2010 Supervisor: Prof. Dr. Ulf Leser Motivation
Proposal for diploma thesis Accurate Long-Read Alignment using Similarity Based Multiple Pattern Alignment and Prefix Tree Indexing Astrid Rheinländer 01-09-2010 Supervisor: Prof. Dr. Ulf Leser Motivation
HISAT2. Fast and sensi0ve alignment against general human popula0on. Daehwan Kim
 HISA2 Fast and sensi0ve alignment against general human popula0on Daehwan Kim infphilo@gmail.com History about BW, FM, XBW, GBW, and GFM BW (1994) BW for Linear path Burrows M, Wheeler DJ: A Block Sor0ng
HISA2 Fast and sensi0ve alignment against general human popula0on Daehwan Kim infphilo@gmail.com History about BW, FM, XBW, GBW, and GFM BW (1994) BW for Linear path Burrows M, Wheeler DJ: A Block Sor0ng
Algorithms in Bioinformatics: A Practical Introduction. Database Search
 Algorithms in Bioinformatics: A Practical Introduction Database Search Biological databases Biological data is double in size every 15 or 16 months Increasing in number of queries: 40,000 queries per day
Algorithms in Bioinformatics: A Practical Introduction Database Search Biological databases Biological data is double in size every 15 or 16 months Increasing in number of queries: 40,000 queries per day
Read Naming Format Specification
 Read Naming Format Specification Karel Břinda Valentina Boeva Gregory Kucherov Version 0.1.3 (4 August 2015) Abstract This document provides a standard for naming simulated Next-Generation Sequencing (Ngs)
Read Naming Format Specification Karel Břinda Valentina Boeva Gregory Kucherov Version 0.1.3 (4 August 2015) Abstract This document provides a standard for naming simulated Next-Generation Sequencing (Ngs)
Sequence comparison: Local alignment
 Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback
Sequence comparison: Local alignment Genome 559: Introuction to Statistical an Computational Genomics Prof. James H. Thomas http://faculty.washington.eu/jht/gs559_217/ Review global alignment en traceback